
25.7: Peptides and Proteins
- Page ID
- 22363

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Classification
Amino acids are the building blocks of the polyamide structures of peptides and proteins. Each amino acid is linked to another by an amide (or peptide) bond formed between the \(\ce{NH_2}\) group of one and the \(\ce{CO_2H}\) group of the other:
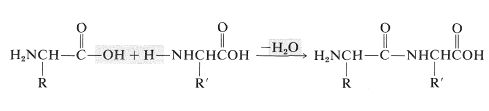
In this manner a polymeric structure of repeating amide links is built into a chain or ring. The amide groups are planar and configuration about the \(\ce{C-N}\) bond is usually, but not always, trans (Section 24-1). The pattern of covalent bonds in a peptide or protein is called its primary structure:
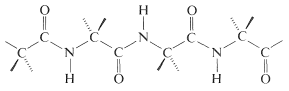
The distinction between a protein and a peptide is not completely clear. One arbitrary choice is to call proteins only those substances with molecular weights greater than 5000. The distinction might also be made in terms of differences in physical properties, particularly hydration and conformation. Thus proteins, in contrast to peptides, have very long chains that are coiled and folded in particular ways, with water molecules filling the voids in the coils and folds. Hydrogen bonding between the amide groups plays a decisive role in holding the chains in juxtaposition to one another, in what is sometimes called the secondary and tertiary structure.\(^5\) Under the influence of heat, organic solvents, salts, and so on, protein molecules undergo changes, often irreversibly, called denaturation. The conformations of the chains and the degree of hydration are thereby altered, with the result that solubility and ability to crystallize decreases. Most importantly, the physiological properties of the protein usually are destroyed permanently on denaturation. Therefore, if a synthesis of a protein is planned, it would be necessary to duplicate not only the amino-acid sequences but also the exact conformations of the chains and the manner of hydration characteristic of the native protein. With peptides, the chemical and physiological properties of natural and synthetic materials usually are identical, provided the synthesis duplicates all of the structural and configurational elements. What this means is that a peptide automatically assumes the secondary and tertiary structure characteristic of the native peptide on crystallization or dissolution in solvents.
Representation of peptide structures of any length with conventional structural formulas is cumbersome. As a result, abbreviations are universally used that employ three-letter symbols for the component amino acids. It is important that you know the conventions for these abbreviations. The two possible dipeptides made up of one glycien and one alanine are
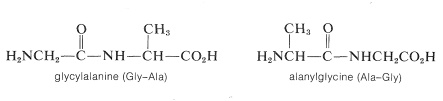
Notice that in the conventions used for names and abbreviated formulas the amino acid with the free amino group (the \(\ce{N}\)-terminal amino acid) always is written on the left. The amino acid with the free carboxyl group (the \(\ce{C}\)-terminal amino acid) always is written on the right. The dash between the three-letter abbreviations for the acids designates that they are linked together by an amide bond.
Determination of Amino-Acid Sequences
The general procedure for determining the primary structure of a peptide or protein consists of three main steps. First, the number and kind of amino-acid units in the primary structure must be determined. Second, the amino acids at the ends of the chains are identified, and third, the sequence of the component amino acids in the chains is determined.
The amino-acid composition usually is obtained by complete acid hydrolysis of the peptide into its component amino acids and analysis of the mixture by ion-exchange chromatography (Section 25-4C). This procedure is complicated by the fact that tryptophan is destroyed under acidic conditions. Also, asparagine and glutamine are converted to aspartic and glutamic acids, respectively.
Determination of the \(\ce{N}\)-terminal acid in the peptide can be made by treatment of the peptide with 2,4-dinitrofluorobenzene, a substance very reactive in nucleophilic displacements with amines but not amides (see Section 14-6B). The product is an \(\ce{N}\)-2,4-dinitrophenyl derivative of the peptide which, after hydrolysis of the amide linkages, produces an \(\ce{N}\)-2,4-dinitrophenylamino acid:
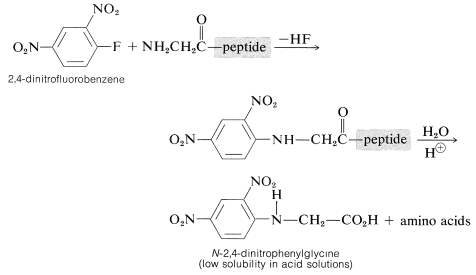
These amino-acid derivatives can be separated from the ordinary amino acids resulting from hydrolysis of the peptide because the low basicity of the 2,4-dinitrophenyl-substituted nitrogen (Section 23-7C) greatly reduces the solubility of the compound in acid solution and alters its chromatographic behavior. The main disadvantage to the method is that the entire peptide must be destroyed in order to identify the one \(\ce{N}\)-terminal acid.
A related and more sensitive method makes a sulfonamide of the terminal \(\ce{NH_2}\) group with a reagent called "dansyl chloride". As with 2,4-dinitrofluorobenzene, the peptide must be destroyed by hydrolysis to release the \(\ce{N}\)-sulfonated amino acid, which can be identified spectroscopically in microgram amounts:

A powerful method of sequencing a peptide from the \(\ce{N}\)-terminal end is the Edman degradation in which phenyl isothiocyanate, \(\ce{C_6H_5N=C=S}\), reacts selectively with the terminal amino acid under mildly basic conditions. If the reaction mixture is then acidified, the terminal amino acid is cleaved from the peptide as a cyclic thiohydantoin, \(8\):
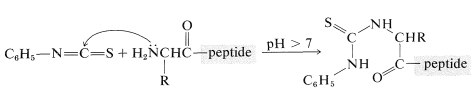
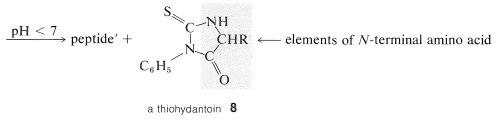
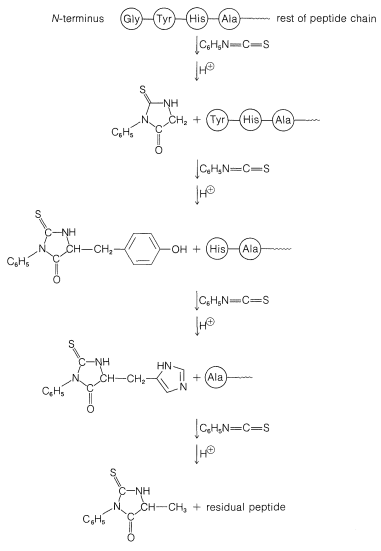
There are simple reagents that react selectively with the carboxyl terminus of a peptide, but they have not proved as generally useful for analysis of the \(\ce{C}\)-terminal amino acids as has the enzyme carboxypeptidase A. This enzyme catalyzes the hydrolysis of the peptide bond connecting the amino acid with the terminal carboxyl groups to the rest of the peptide. Thus the amino acids at the carboxyl end will be removed one by one through the action of the enzyme. Provided that appropriate corrections are made for different rates of hydrolysis of peptide bonds for different amino acids at the carboxyl end of the peptide, the sequence of up to five or six amino acids in the peptide can be deduced from the order of their release by carboxypeptidase. Thus a sequence such as peptide-Ser-Leu-Tyr could be established by observing that carboxypeptidase releases amino acids from the peptide in the order Tyr, Leu, Ser:

Determining the amino-acid sequences of large peptides and proteins is very difficult. Although the Edman degradation and even carboxypeptidase can be used to completely sequence small peptides, they cannot be applied successfully to peptide chains with several hundred amino acid units. Success has been obtained with long peptide chains by employing reagents, often enzymes, to selectively cleave certain peptide bonds. In this way the chain can be broken down into several smaller peptides that can be separated and sequenced. The problem then is to determine the sequence of these small peptides in the original structure. To do this, alternative procedures for selective cleavages are carried out that produce different sets of smaller peptides. It is not usually necessary to sequence completely all of the peptide sets. The overall amino-acid composition and the respective end groups of each peptide may suffice to show overlapping sequences from which the complete amino-acid sequence logically can be deduced.
The best way to show you how the overlap method of peptide sequencing works is by a specific example. In this example, we will illustrate the use of the two most commonly used enzymes for selective peptide cleavage. One is trypsin, a proteolytic enzyme of the pancreas (MW 24,000) that selectively catalyzes the hydrolysis of the peptide bonds of basic amino acids, lysine and arginine. Cleavage occurs on the carboxyl side of lysine or arginine:
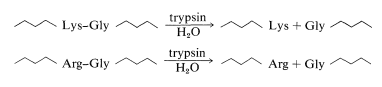
Chymotrypsin is a proteolytic enzyme of the pancreas (MW 24,500) that catalyzes the hydrolysis of peptide bonds to the aromatic amino acids, tyrosine, tryptophan, and phenylalanine, more rapidly than to other amino acids. Cleavage occurs on the carboxyl side of the aromatic amino acid:
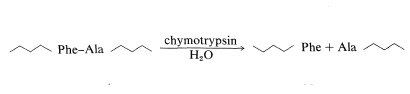
Our example is the sequencing of a peptide (P) derived from partial hydrolysis of a protein which, on complete acid hydrolysis, gave Ala, 3 Gly, Glu, His, 3 Lys, Phe, Tyr, 2 Val, and one molar equivalent of ammonia.
1. Treatment of the peptide (P) with carboxypeptidase released alanine, and with 2,4-dinitrofluorobenzene followed by hydrolysis gave the 2,4-dinitrophenyl derivative of valine. These results establish the \(\ce{N}\)-terminus as valine and the \(\ce{C}\)-terminus as alanine. The known structural elements now are
2. Partial hydrolysis of the peptide (P) with trypsin gave a hexapeptide, a tetrapeptide, a dipeptide, and one molar equivalent of lysine. The peptides, which we will designated respectively as M, N, and O, were sequenced by Edman degradation and found to have structures:
\[\begin{array}{ll} \text{Gly}-\text{Ala} & \text{O} \\ \text{Val}-\text{Tyr}-\text{Glu}-\text{Lys} & \text{N} \\ \text{Val}-\text{Gly}-\text{Phe}-\text{Gly}-\text{His}-\text{Lys} & \text{M} \end{array}\]
With this information, four possible structures can be written for the original peptide P that are consistent with the known end groups and the fact that trypsin cleaves the peptide P on the carboxyl side of the lysine unit. Thus
\[\begin{array}{cc} \text{N}-\text{M}-\text{Lys}-\text{O} & \text{M}-\text{N}-\text{Lys}-\text{O} \\ \text{N}-\text{Lys}-\text{M}-\text{O} & \text{M}-\text{Lys}-\text{N}-\text{O} \end{array}\]
3. Partial hydrolysis of the peptide P using chymotrypsin as catalyst gave three peptides, X, Y, and Z. These were not sequenced, but their amino-acid composition was determined:
\[\begin{array}{ll} \text{Gly, Phe, Val} & \text{X} \\ \text{Gly, His, Lys, Tyr, Val} & \text{Y} \\ \text{Ala, Glu, Gly, 2 Lys} & \text{Z} \end{array}\]
This information can be used to decide which of the alternative structures deduced above is correct. Chymotrypsin cleaves the peptide on the carboxyl side of the phenylalanine and tyrosine units. Only peptide M contains Phe, and if we compare M with the compositions of X, Y, and Z, we see that only X and Y overlap with M. Peptide Z contains the only Ala unit and must be the \(\ce{C}\)-terminus. If we put together these pieces to get a peptide, P' (which differs from P by not having the nitrogen corresponding to the ammonia formed on complete hydrolysis) then P' must have the structure X-Y-Z:
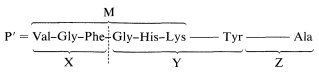
This may not be completely clear, and it will be well to consider the logic in some detail. Peptides M and N both have \(\ce{N}\)-terminal valines, and one of them must be the \(\ce{N}\)-terminal unit. Peptide M overlaps with X and Y, and because X and Y are produced by a cleavage on the carboxyl side of Phe, the X and Y units have to be connected in the order X-Y. Because the other Val is in Y, the \(\ce{N}\)-terminus must be M. This narrows the possibilities to
\[\begin{array}{c} \text{M}-\text{N}-\text{Lys}-\text{O} \\ \text{M}-\text{Lys}-\text{N}-\text{O} \end{array}\]
There are two Lys units in Z, and this means that only the sequence M-N-Lys-O is consistent with the sequence X-Y-Z, as shown:
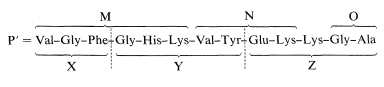
The final piece of the puzzle is the placement of the mole of ammonia released from the original peptide on acid hydrolysis. The ammonia comes from a primary amide function:
\[\ce{R-CONH_2} \overset{\ce{H_3O}^\oplus}{\longrightarrow} \ce{RCO_2H} + \ce{NH_4^-}\]
The amide group cannot be at the \(\ce{C}\)-terminus because the peptide would then be inert to carboxypeptidase. The only other possible place is on the side-chain carboxyl of glutamic acid. The complete structure may be written as
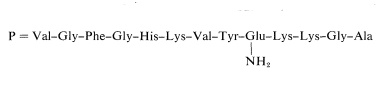
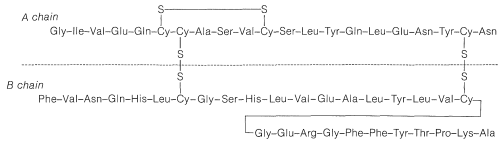
Using procedures such as those outlined in this section more than 100 proteins have been sequenced. This is an impressive accomplishment considering the complexity and size of many of these molecules (see, for example, Table 25-3). It has been little more than two decades since the first amino acid sequence of a protein was reported by F. Sanger, who determined the primary structure of insulin (1953). This work remains a landmark in the history of chemistry because it established for the first time that proteins have definite primary structures in the same way that other organic molecules do. Up until that time, the concept of definite primary structures for proteins was openly questioned. Sanger developed the method of analysis for \(\ce{N}\)-terminal amino acids using 2,4-dinitrofluorobenzene and received a Nobel Prize in 1958 for his success in determining the amino-acid sequence of insulin.
Methods for Forming Peptide Bonds
The problems involved in peptide syntheses are of much practical importance and have received considerable attention. The major difficulty in putting together a chain of say 100 amino acids in a particular order is one of overall yield. At least 100 separate synthetic steps would be required, and, if the yield in each step were equal to \(n \times 100\%\), the overall yield would be \(\left( n^{100} \times 100\% \right)\). If the yield in each step were \(90\%\), the overall yield would be only \(0.003\%\). Obviously, a practical laboratory synthesis of a peptide chain must be a highly efficient process. The extraordinary ability of living cells to achieve syntheses of this nature, not of just one but of a wide variety of such substances, is truly impressive.
Several methods for the formation of amide bonds have been discussed in Sections 18-7A and 24-3A. The most general reaction is shown below, in which X is some reactive leaving group (see Table 24-1):
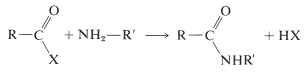
When applied to coupling two different amino acids, difficulty is to be expected because these same reactions can link two amino acids in a total of four different ways. Thus if we started with a mixture of glycine and alanine, we could generate for dipeptides, Gly-Ala, Ala-Gly, Gly-Gly, and Ala-Ala.
To avoid unwanted coupling reactions a protecting group is substituted on the amino function of the acid that is to act as the acylating agent. Furthermore, all of the amino, hydroxyl, and thiol functions that may be acylated to give undesired products usually must be protected. For instance, to synthesize Gly-Ala free of other possible dipeptides, we would have to protect the amino group of glycine and the carboxyl group of alanine:
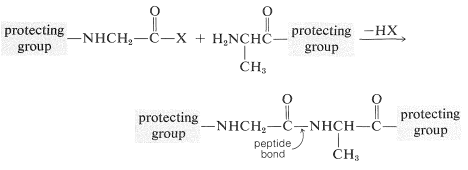
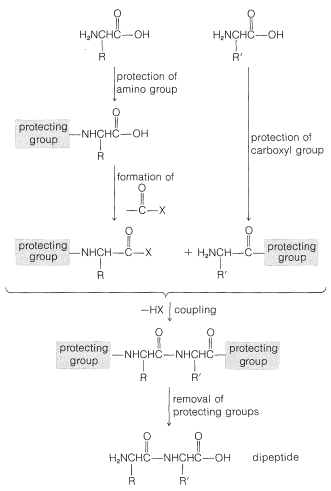
Some methods of protecting amine and hydroxyl functions were discussed previously in Sections 23-13 and 15-9, respectively. A summary of some commonly used protecting groups for \(\ce{NH_2}\), \(\ce{OH}\), \(\ce{SH}\), and \(\ce{CO_2H}\) functions is in Table 25-2, together with the conditions by which the protecting groups may be removed. The best protecting groups for \(\ce{NH_2}\) functions are phenylmethoxycarbonyl (benzyloxycarbonyl) and tert-butoxycarbonyl. Both groups can be removed by treatment with acid, although the tert-butoxycarbonyl group is more reactive. The phenylmethoxycarbonyl group can be removed by reduction with either hydrogen over a metal catalyst or with sodium in liquid ammonia. This method is most useful when, in the removal step, it is necessary to avoid treatment with acid:
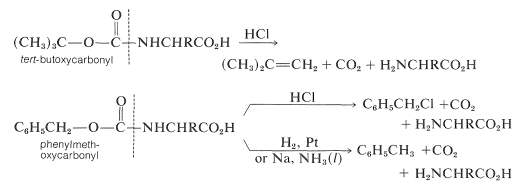
Table 25-2: Some Amine and Carboxyl Protecting Groups Used in Peptide Syntheses
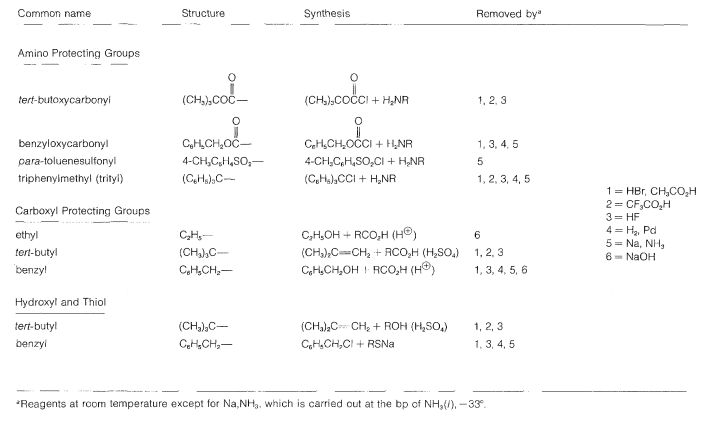
In most cases, formation of the ethyl ester provides a satisfactory protecting group for the carboxyl function.
Conversion of the carboxyl group to a more reactive group and coupling are key steps in peptide synthesis. The coupling reaction must occur readily and quantitatively, and with a minimum of racemization of the chiral centers in the molecule. This last criterion is the Achilles' heel of many possible coupling sequences. The importance of nonracemization can best be appreciated by an example. Consider synthesis of a tripeptide from three protected \(L\)-amino acids, A, B, and C, in two sequential coupling steps, \(\text{C} \overset{\text{B}}{\rightarrow} \text{B}-\text{C} \overset{\text{A}}{\rightarrow} \text{A}-\text{B}-\text{C}\). Suppose that the coupling yield is quantitative, but there is \(20\%\) formation of the \(D\) isomer in the acylating component in each coupling step. Then the tripeptide will consist of a mixture of four diastereomers, only \(64\%\) of which will be the desired \(L\),\(L\),\(L\) diastereomer (Equation 25-7):
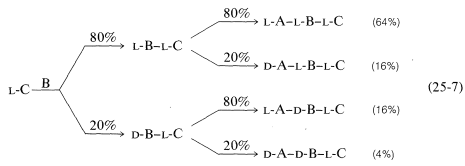 \(\tag{25-7}\)
\(\tag{25-7}\)
This is clearly unacceptable, especially for longer-chain peptides. Nine coupling steps with \(20\%\) of the wrong isomer formed in each would give only \(13\%\) of the decapeptide with the correct stereochemistry.
The most frequently used carboxyl derivatives in amide coupling are azides, \(\ce{RCO-N_3}\), mixed anhydrides, \(\ce{RCO-O-COR'}\), and esters of moderately acidic phenols, \(\ce{RCO-OAr}\) (see Table 24-1). It also is possible to couple free acid with an amine group using a diimide, \(\ce{R-N=C=N-R}\), most frequently \(\ce{N}\),\(\ce{N'}\)-dicyclohexylcarbodiimide.
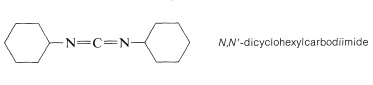
The diimide reagent may be thought of as a dehydrating agent. The "elements of water" eliminated in the coupling are consumed by the diimide to form a substituted urea. The overall reaction is
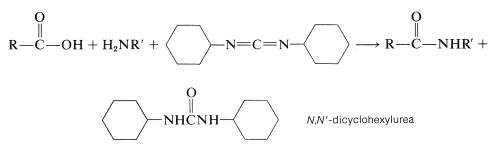
This reaction takes place because diimides, \(\ce{-N=C=N}-\), have reactive cumulated double-bond systems like those of ketenes, \(\ce{-C=C=O}\); isocyanates, \(\ce{-N=C=O}\); and isothiocyanates, \(\ce{-N=C=S}\); and are susceptible to nucleophilic attack at the central carbon. In the first step of the diimide-coupling reaction, the carboxyl function adds to the imide to give an acyl intermediate, \(9\). This intermediate is an activated carboxyl derivative \(\ce{RCO-X}\) and is much more reactive toward an amino function than is the parent acid. The second step therefore is the aminolysis of \(9\) to give the coupled product and \(\ce{N}\),\(\ce{N'}\)-dicyclohexylurea:
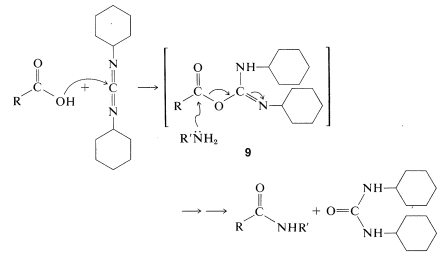
After completion of a coupling reaction, and before another amino acid can be added to the \(\ce{N}\)-terminus, it is necessary to remove the protecting group. This must be done by selective reactions that do not destroy the peptide bonds or side-chain protecting groups. This part of peptide synthesis is discussed in Section 23-13, and some reactions useful for removal of the \(\ce{N}\)-terminal protecting groups are summarized in Table 25-2.
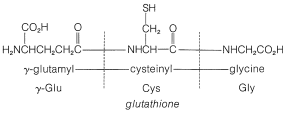
In spite of the large number of independent steps involved in the synthesis of even small peptides, each with its attendant problems of yield, racemization, and selectivity, remarkable success has been achieved in the synthesis of large peptides and certain of the smaller peptides. The synthesis of insulin (Figure 25-8) with its 51 amino acid units and 3-disulfide bridges has been achieved by several investigators. Several important hormonal peptides, namely glutathione, oxytocin, vasopressin, and thyrotropic hormone (see Figure 25-9) have been synthesized. A major accomplishment has been the synthesis of an enzyme with ribonuclease activity reported independently by two groups of investigators, led by R. Hirschman (Merck) and R. B. Merrifield (Rockefeller University). This enzyme is one of the simpler proteins, having a linear stricture of 124 amino-acid residues. It is like a peptide, not a protein, in that it assumes the appropriate secondary and tertiary structure without biochemical intervention (Section 25-7A). As a specific example of the strategy involved in peptide synthesis, the stepwise synthesis of oxytocin is outlined in Figure 25-10, using the abbreviated notation in common usage.
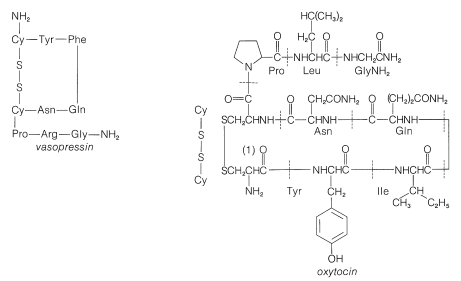
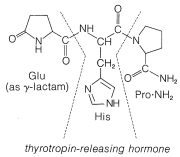
not to \(\ce{C_1}\). Vasopressin (middle left) and oxytocin (middle right) are peptide hormones from the posterior lobe of the pituitary gland. They function primarily to raise blood pressure (vasopressin), as antidiuretic (vasopressin), and to promote contraction of uterus and lactation muscles (oxytocin). The isolation, identification, and synthesis of these hormones was accomplished by Vincent du Vigneaud, for which he was awarded the Nobel Prize in chemistry in 1965. Thyrotropin-releasing hormone (bottom) is one of several small peptide hormones secreted by the anterior lobe of the pituitary gland. These are the "master" hormones that function to stimulate hormone secretion from other endocrine glands. Thyrotropin stimulates the functioning of the thyroid gland.
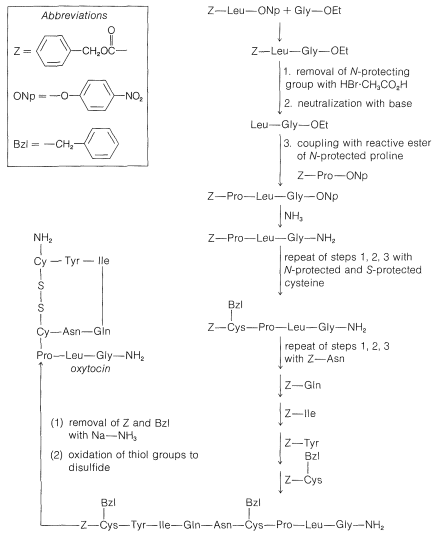
Figure 25-10: Stepwise synthesis of oxytocin by the reactive ester method. In the abbreviations used here \(-\text{Gly}-\ce{NH_2} = \ce{-NHCH_2CONH_2}\) and
Solid-Phase Peptide Synthesis
The overall yield in a multistep synthesis of a peptide of even modest size is very poor unless each step can be carried out very efficiently. An elegant modification of classical peptide synthesis has been developed by R. B. Merrifield, which offers improved yields by minimizing manipulative losses that normally attend each step of a multistage synthesis. The key innovation is to anchor the \(\ce{C}\)-terminal amino acid to an insoluble support, and then add amino-acid units by the methods used for solution syntheses. After the desired sequence of amino acids has been achieved, the peptide can be cleaved from the support and recovered from solution. All the reactions involved in the synthesis must, of course, be brought to essentially \(100\%\) completion so that a homogeneous product can be obtained. The advantage of having the peptide anchored to a solid support is that laborious purification steps are virtually eliminated; solid material is purified simply by washing and filtering without transferring the material from one container to another. The method has become known as solid-phase peptide synthesis. More of the details of the solid-phase synthesis follow.
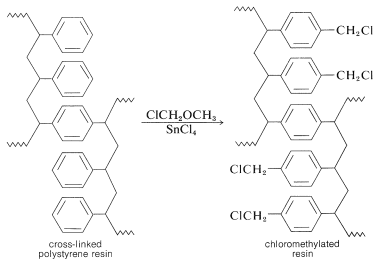
The nature of the polymer support is of great importance for a successful peptide synthesis. One that is widely used is a cross-linked polystyrene resin of the type employed in ion-exchange chromatography (Section 25-4C). It is necessary that the resin be insoluble but have a loose enough structure to absorb organic solvents. Otherwise, the reagents will not be able to penetrate into the spaces between the chains. This is undesirable because the reactions occur on the surface of the resin particles and poor penetration greatly reduces the number of equivalents of reactive sites that can be obtained per gram of resin. Finally, to anchor a peptide chain to the resin, a reactive functional group (usually a chloromethyl group) must be introduced into the resin. This can be done by a Friedel-Crafts chloromethylation reaction, which substitutes the \(\ce{ClCH_2}-\) group in the 4-position of the phenyl groups in the resin:
At the start of the peptide synthesis, the \(\ce{C}\)-terminal amino acid is bonded through its carboxyl group to the resin by a nucleophilic attack of the carboxylate ion on the chloromethyl groups. The \(\alpha\)-amino group must be suitably protected, as with tert-butoxycarbonyl, before carrying out this step:
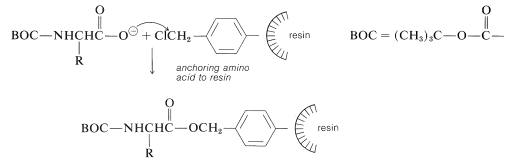
Next, the amine protecting group must be removed without cleaving the ester bond to the resin. The coupling step to a second \(\ce{N}\)-protected amino acid follows, with \(\ce{N}\),\(\ce{N'}\)-dicyclohexylcarbodiimide as the coupling reagent of choice:
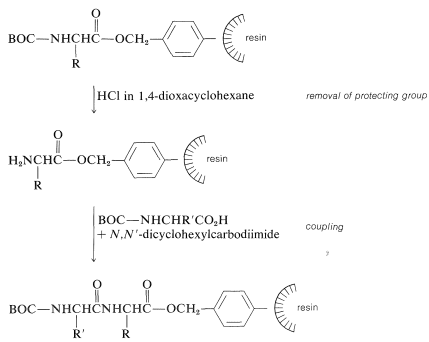
The peptide-bond-forming steps are repeated as many times as needed to build up the desired sequence. Ultimately, the peptide chain is removed from the resin, usually with \(\ce{HBr}\) in anhydrous trifluoroethanoic acid, \(\ce{CF_3CO_2H}\), or with anhydrous \(\ce{HF}\). This treatment also removes the other acid-sensitive protecting groups.
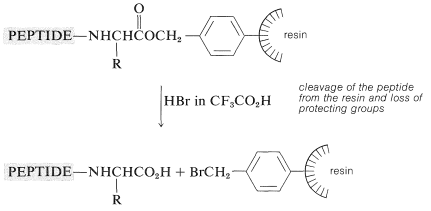
The method lends itself beautifully to automatic control, and machines suitably programmed to add reagents and wash the product at appropriate times have been developed. At present, the chain can be extended by six or so amino acid units a day. It is necessary to check the homogeneity of the growing peptide chain at intervals because if any step does not proceed properly, the final product can be seriously contaminated with peptides with the wrong sequence.
In the synthesis of the enzyme ribonuclease by the Merrifield method, the 124 amino acids were arranged in the ribonuclease sequence through 369 reactions and some 12,000 individual operations of the automated peptide-synthesis machine without isolation of any intermediates.
Separation of Peptides and Proteins
In many problems of peptide sequencing and peptide synthesis it is necessary to be able to separate mixtures of peptides and proteins. The principal methods used for this purpose depend on acid-base properties or on molecular sizes and shapes.
Ultracentrifugation is widely used for the purification, separation, and molecular-weight determination of proteins. A centrifugal field, up to 500,000 times that of gravity, is applied to the solution, and molecules move downward in the field according to their mass and size.
Large molecules also can be separated by gel filtration (or gel chromatography), wherein small molecules are separated from large ones by passing a solution over a gel that has pores of a size that the small molecules can penetrate into and be trapped. Molecules larger than the pore size are carried on with the solvent. This form of chromatographic separation is based on "sieving" rather than on chemical affinity. A wide range of gels with different pore sizes is available, and it is possible to fractionate molecules with molecular weights ranging from 700 to 200,000. The molecular weight of a protein can be estimated by the sizes of the pores that it will, or will not, penetrate.
The acid-base properties, and hence ionic character, of peptides and proteins also can be used to achieve separations. Ion-exchange chromatography, similar to that described for amino acids (Section 25-4C), is an important separation method. Another method based on acid-base character and molecular size depends on differential rates of migration of the ionized forms of a protein in an electric field (electrophoresis). Proteins, like amino acids, have isoelectric points, which are the pH values at which the molecules have no net charge. At all other pH values there will be some degree of net ionic charge. Because different proteins have different ionic properties, they frequently can be separated by electrophoresis in buffered solutions. Another method, which is used for the separation and purification of enzymes, is affinity chromatography, which was described briefly in Section 9-2B.
\(^5\) The distinction between secondary and tertiary structure is not sharp. Secondary structure involves consideration of the interactions and spatial relationships of the amino acids in the peptide chains that are close together in the primary structure, whereas tertiary structure is concerned with those that are far apart in the primary structure.
Contributors and Attributions
John D. Robert and Marjorie C. Caserio (1977) Basic Principles of Organic Chemistry, second edition. W. A. Benjamin, Inc. , Menlo Park, CA. ISBN 0-8053-8329-8. This content is copyrighted under the following conditions, "You are granted permission for individual, educational, research and non-commercial reproduction, distribution, display and performance of this work in any format."