
9.3: Chromatographic Separation Procedures
- Page ID
- 22156

Gas-Liquid Chromatography
Many separation methods are based on chromatography, that is, separation of the components of a mixture by differences in the way they become distributed (or partitioned) between two different phases. To illustrate with an extreme example, suppose we have a mixture of gaseous methane and ammonia and contact this mixture with water. Ammonia, being very soluble in water (~\(90 \: \text{g}\) per \(100 \: \text{g}\) water at \(1 \: \text{atm}\) pressure), will mostly go into the water phase, whereas the methane, being almost insoluble (~\(0.003 \: \text{g}\) per \(100 \: \text{g}\) of water) will essentially remain entirely in the gas phase. Such a separation of methane and ammonia would be a one-stage partitioning between gas and liquid phases and, clearly, could be made much more efficient by contacting the gas layer repeatedly with fresh water. Carried through many separate operations, this partitioning procedure is, at best, a tedious process, especially if the compounds to be separated are similar in their distributions between the phases. However, partitioning can be achieved nearly automatically by using chromatographic columns, which permit a stationary phase to be contacted by a moving phase. To illustrate, suppose a sample of a gaseous mixture of ammonia and methane is injected into a long tube (column) filled with glass beads moistened with water (the stationary phase), and a slow stream of an inert carrier gas, such as nitrogen or helium, is passed in to push the other gases through. A multistage partitioning would occur as the ammonia dissolves in the water and the resulting gas stream encounters fresh water as it moves along the column. Carrier gas enriched with methane would emerge first and effluent gas containing ammonia would come out later. This is a crude description of the method of gas-liquid chromatography (abbreviated often as glc, GC, or called vapor-phase chromatography, vpc). This technique has become so efficient as to revolutionize the analysis and separation of almost any organic substance that has even a slight degree of volatility at some reasonably attainable temperature. The most modern glc equipment runs wholly under computer control, with preprogrammed temperatures and digital integration of the detector output. A wide variety of schemes is available for measuring the concentration of materials in the effluent carrier gas, and some of these are of such extraordinary sensitivity that only very small samples are necessary (\(10^{-9} \: \text{g}\), or less).
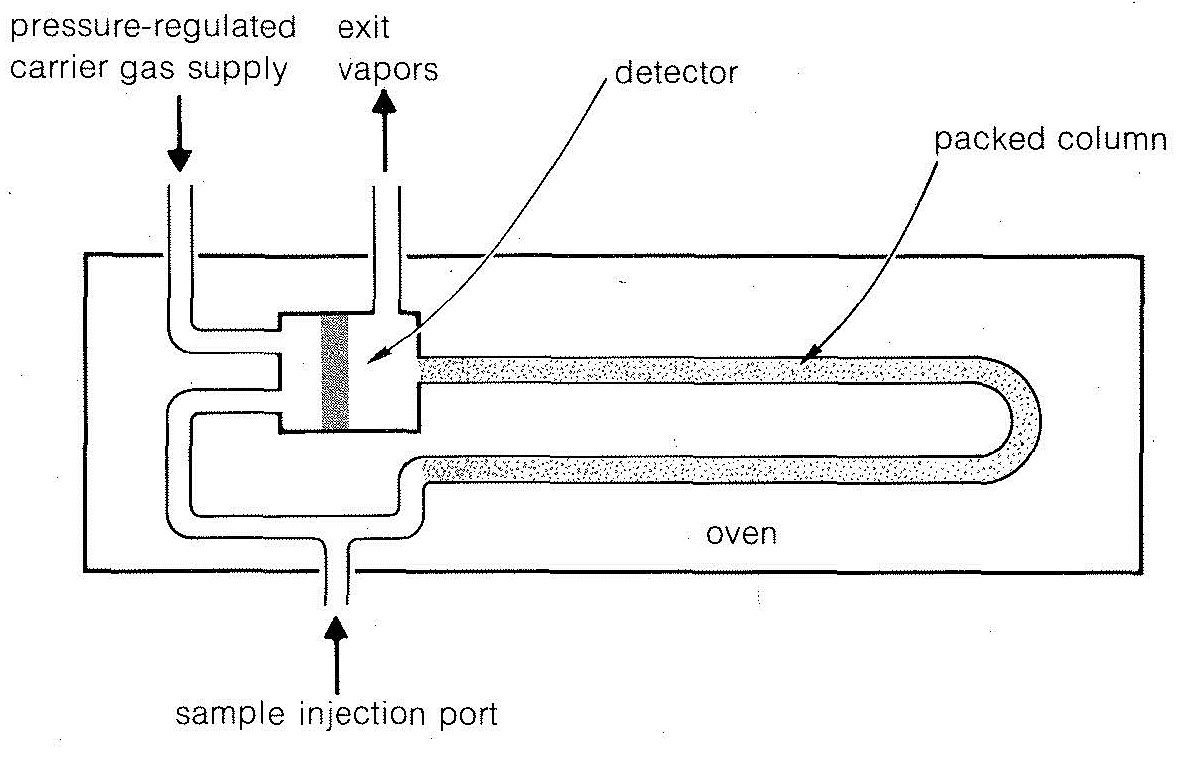
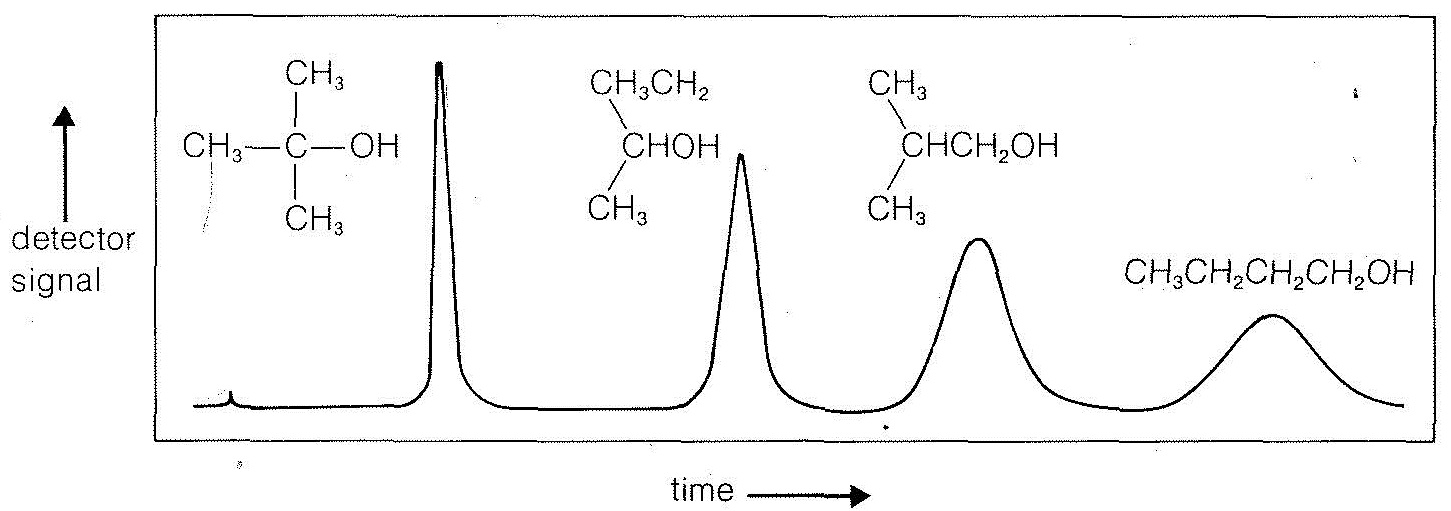
In the usual glc procedure, a few microliters of an organic liquid to be analyzed are injected into a vaporizer and carried with a stream of gas (usually helium) into a long heated column that is packed with a porous solid (such as crushed firebrick) impregnated with a nonvolatile liquid. Gas-liquid partitioning occurs, and small differences between partitioning of the components can be magnified by the large number of repetitive partitions possible in a long column. Detection often is achieved simply by measuring changes in thermal conductivity of the effluent gases. A schematic diagram of the apparatus and a typical separation pattern are shown in Figures 9-1 and 9-2. The method is extraordinarily useful for detection of minute amounts of impurities provided these are separated from the main peak. Glc also can be used effectively to purify materials as well as to detect impurities. To do this, the sample size and the size of the apparatus may be increased, or an automatic system may be used wherein the products from many small-scale runs are combined.
Liquid-Solid Chromatography
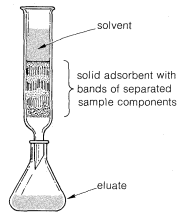
Liquid-solid chromatography originally was developed for the separation of colored substances, hence the name chromatography, which stems from the Greek word chroma meaning color. In a typical examination, a colored substance suspected of containing colored impurities is dissolved in a suitable solvent and the solution allowed to percolate down through a column packed with a solid adsorbent, such as alumina or silica, as shown in Figure 9-3. The "chromatogram" then is "developed" by passing through a suitable solvent that washes the adsorbate down through the column. What one hopes for, but may not always find, is that the components of the mixture will be adsorbed unequally by the solid phase so distinct bands or zones of color appear. The bands at the top of the column contain the most strongly adsorbed components and the bands at the bottom the least strongly held components. The zones may be separated mechanically, or sufficient solvent can be added to wash, or elute, the zones of adsorbed materials sequentially from the column for further analysis.
Liquid-solid chromatography in the form just described was developed first by the Russian biochemist M. S. Tswett, about 1906. In recent years, many variations have been developed that provide greater convenience, better separating power, and wider applicability. In thin-layer chromatography, which is especially useful for rapid analyses, a solid adsorbent containing a suitable binder is spread evenly on a glass plate, a drop of solution to be analyzed is placed near one edge and the plate is placed in a container with the edge of the plate below the spot, dipping into an eluting solvent. The solvent ascends the plate and the materials in the spot move upward at different rates, as on a Tswett column. Various detecting means are used - simple visual observation for colored compounds, differential fluorescence under ultraviolet light, and spraying of the plate with substances that will give colored materials with the compounds present. In favorable cases, this form of liquid-solid chromatography can be carried out with submicrogram quantities of materials.
An extremely important improvement on the Tswett procedure is high-pressure solid-liquid chromatography (HPLC). Increasing the input pressure on the system to \(20\)-\(70 \: \text{atm}\) improves the speed of separations by permitting the use of much smaller solid particles (with more surface area) than would be practical for gravity-flow Tswett columns. Automatic monitoring of the column effluent by ultraviolet spectroscopy (Section 9-9) or by changes in the refractive index usually provides an effective means of determining how the separation is proceeding. With such techniques chromatograms similar to Figure 9-2 are obtained. High-pressure liquid chromatography (hplc) has great advantages for analysis and separation of high-molecular-weight heat-sensitive compounds that are unsuitable for glc.
An ingenious variation of solid-liquid chromatography is to use a solid support to which material is attached that has a specific affinity for a particular substance to be separated. The technique is especially useful for separating enzymes, and the immobile phase can be constructed from compounds known to react with, or be complexed by, the enzyme. Some other forms of chromatography are discussed in Sections 25-4B and 25-7E.
Observation of a single peak in a given chromatographic procedure is evidence, albeit not definitive evidence, for purity. Contaminants with nearly the same properties may be very difficult to separate and, if knowing the degree of purity is highly important, one can run chromatograms with a variety of different adsorbents to see if each gives the same result. If they do, the presumption of purity improves, although it is desirable to determine whether the spectroscopic techniques to be described in the following section permit the same conclusion.
References
John D. Robert and Marjorie C. Caserio (1977) Basic Principles of Organic Chemistry, second edition. W. A. Benjamin, Inc. , Menlo Park, CA. ISBN 0-8053-8329-8. This content is copyrighted under the following conditions, "You are granted permission for individual, educational, research and non-commercial reproduction, distribution, display and performance of this work in any format."