
6.4: Integrated Rate Laws
- Page ID
- 499147

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)- Explain the form and function of an integrated rate law
- Perform integrated rate law calculations for zero-, first-, and second-order reactions
- Define half-life and carry out related calculations
- Identify the order of a reaction by plotting concentration vs. time data according to expected reaction orders
The rate laws we have seen so far relate reaction rates to reactant concentrations. Using calculus, the rate law can be integrated to derive an equation that relates reactant concentration to time. This can be done for any reaction order, but in this chapter we will focus on the integrated rate laws for zero-, first-, and second-order reactions.
Using the integrated rate law equations we can:
- Determine how much reactant or product is present after a given time.
- Estimate how long a reaction takes to reach a certain extent.
- Determine the reaction order of a reactant by plotting the change in concentration over time using the expected linear relationships for zero-, first-, and second-order reactions
Zero-Order Integrated Rate Law
A zero-order reaction is one in which the reaction rate is independent of reactant concentration. This means that the reaction proceeds at a constant rate, regardless of how much reactant is present. The rate law for a zero-order reaction is:
\[\ce{Rate}=-\dfrac{\Delta [A]}{\Delta t}=k[A]^0=k \nonumber \]
CHM135 does not require you to use calculus on assessments. However, we will derive the integrated rate laws here so you understand where they come from, after that you can simply use them. As shown above, the rate of a zero order reaction is independent of concentration:
\[-\dfrac{d[A]}{dt}=k \nonumber\]
This equation can be rearranged to:
\[ d[A] =-k \: dt \nonumber\]
By separating our variables and integrating our rate laws with respect \([A]\) on the left from \([A]_o\) to \([A]_t\) and with respect to \(t\) on the right from 0 to t,
\[\int\limits_{[A]_o}^{[A]_t} \, d[A] = \int\limits_{0}^{t} -k\, dt \nonumber\]
we can generate the following linear relationships between concentration and time:
\[ [A]−[A]_0=−kt \nonumber\]
This is the integrated rate equation for a zero-order reaction, but it is typically rearranged to the following format:
\[ [A]=−kt+[A]_0 \nonumber\]
As it follows the general form of a linear equation:
\[ y =mx+b \notag \]
As shown in Figure \(\PageIndex{1}\) the plot of concentration versus time for a zero-order reaction is a straight line with a slope of −k and an intercept of [A]0.

Figure \(\PageIndex{1}\) shows a plot of concentration versus time for a zero order reaction.
If the reaction order is known to be zero then we can use the integrated rate equation above to calculate changes in concentration over time. If the reaction order is unknown we can test whether a reaction is zero-order by plotting concentration versus time as a reaction proceeds. If this gives a linear relationship then the reaction order for that reactant is zero. When we do this we are modeling the reaction as if it were zero-order and seeing how the data fits this model. If it's not linear then we need to explore another model (maybe first-order or second-order).
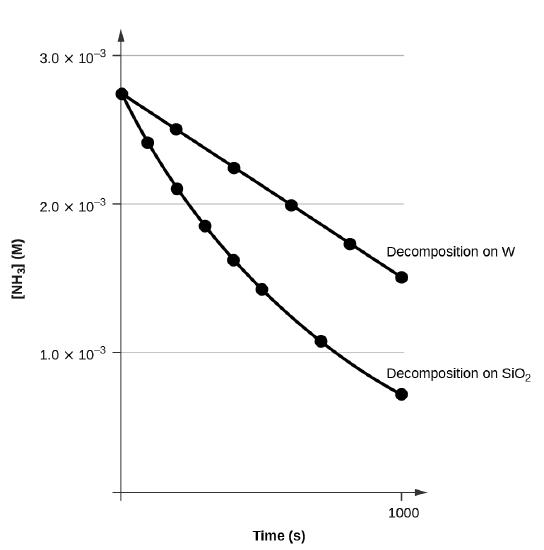
Figure \(\PageIndex{2}\) shows an example of this approach where experimental data for the decomposition of ammonia (NH3) in the presence of two different materials at high temperatures are plotted together:
- Decomposition of ammonia (NH3) on tungsten (W)
- Decomposition of ammonia (NH3) on silica (SiO2)
From the plot of [NH3] versus time we can see that the decomposition on tungsten gives a straight line while that on silica does not, indicating the reaction order for NH3 is zero when it involves tungsten but the reaction order of NH3 is not zero when it involves silica.
Having a reaction order of zero feels a bit strange, but this is not unusual when a reaction involves a solid catalyst, which is the case here. Ammonia gas diffuses onto the surface of the solid tungsten first and then once there they react. Once the surface is fully occupied, adding more ammonia does not increase the rate, leading to zero-order kinetics. From the slope of the line for the zero-order decomposition, the rate constant can be determined:
\[\ce{slope}=−k=\mathrm{1.3110^{−6}\:mol/L/s} \nonumber \]
First-Order Integrated Rate Law
The rate law for a first-order reaction is:
\[\ce{Rate}=-\dfrac{\Delta [A]}{\Delta t}=k[A]^1 \nonumber \]
Changing deltas to differentials,
\[\ -\dfrac{d[A]}{dt} =k [A] \nonumber \]
then rearranging the equation to separate the variables \([A]\) and \(t\),
\[\ \dfrac{1}{[A]} d[A]=-k \: dt \nonumber \]
we can integrate with respect to \([A]\) on the left from \([A]_o\) to \([A]_t\) and with respect to \(t\) on the right from 0 to t:
\[\int\limits_{[A]_o}^{[A]_t} \dfrac{1}{[A]}\, d[A] = \int\limits_{0}^{t} -k\, dt \nonumber\]
This gives us the first-order integrated rate law, which provides a linear expression for changes in reactant concentration as a function of time,
\[ ln[A]-\ln[A]_0=−kt \nonumber\]
or,
\[ \ln\left(\dfrac{[A]_t}{[A]_0}\right)=−kt \nonumber\]
which can be rearranged into the form of a linear equation:
\[ ln[A]=−kt+\ln[A]_0 \nonumber\]
\[ y=mx+b \notag \]
This is the integrated rate expression for a first-order reaction, and, similar to the zero-order reactions in the section above, this equation allows us to calculate the concentration of A at any time ([A]t), given the initial concentration ([A]0), the rate constant (k) and the elapsed time (t).
For a first-order reaction, a plot of \(ln[A]\) versus \(t\) yields a straight line with a slope of \(−k\) and an intercept of \( ln[A]_0\). If data plotted this way does not result in a straight line, the reaction is not first order in \(A\).
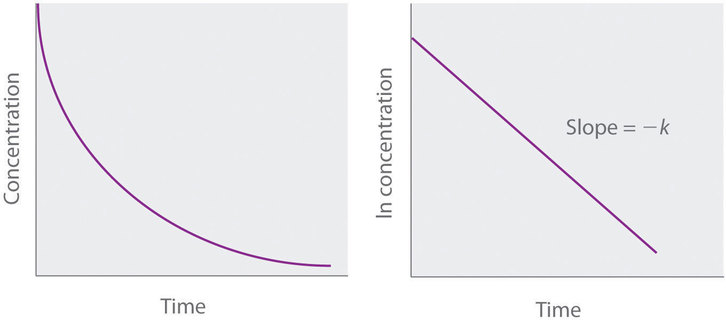
Figure \(\PageIndex{3}\): Graphs of a first-order reaction. The expected shapes of the curves for plots of reactant concentration versus time (left) and the natural logarithm of reactant concentration versus time (right) for a first-order reaction.
The rate constant for the first-order decomposition of cyclobutane, \(\ce{C4H8}\) at 500 °C is 9.2 × 10−3 s−1:
\[\ce{C4H8⟶2C2H4} \nonumber \]
How long will it take for 80.0% of a sample of C4H8 to decompose?
Solution
Since we need to find time, we use the integrated form of the rate law :
\[\ln\left(\dfrac{[A]_0}{[A]}\right)=kt \nonumber \]
The initial concentration of C4H8, [A]0, is not provided, we know that 80.0% of the sample has decomposed,, meaning that 20.0% remains:
[A]t = 0.200[A]0
Rearranging the rate law to isolate t and substituting the provided quantities yields:
\[\begin{align*}
t&=\ln\dfrac{[A]_0}{[0.200[A]_0]}×\dfrac{1}{k}\\[4pt]
&=\mathrm{\ln\dfrac{1}{0.200}×\dfrac{1}{9.2×10^{−3}\:s^{−1}}}\\[4pt]
&=\mathrm{1.609×\dfrac{1}{9.2×10^{−3}\:s^{−1}}}\\[4pt]
&=\mathrm{1.7×10^2\:s}
\end{align*} \nonumber \]
Iodine-131 is a radioactive isotope that is used to diagnose and treat some forms of thyroid cancer. Iodine-131 decays to xenon-131 according to the equation:
\[\textrm{I-131 ⟶ Xe-131 + electron} \nonumber \]
The decay is first-order with a rate constant of 0.138 d−1. All radioactive decay is first order. How many days will it take for 90% of the iodine−131 in a 0.500 M solution of this substance to decay to Xe-131?
- Answer
-
16.7 days
Demonstrate that the decomposition of H2O2 follows a first-order rate law by graphing ln[H2O2] versus time. Determine the rate constant for the rate of decomposition of H2O2.
| Time (h) | [H2O2] (M) | ln[H2O2] |
|---|---|---|
| 0 | 1.000 | 0.0 |
| 6.00 | 0.500 | −0.693 |
| 12.00 | 0.250 | −1.386 |
| 18.00 | 0.125 | −2.079 |
| 24.00 | 0.0625 | −2.772 |
Solution
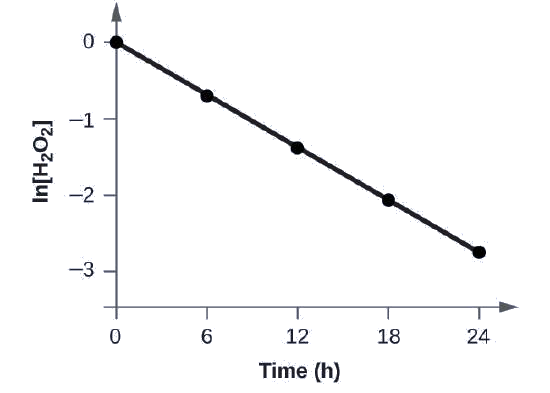
The linear relationship between the ln[H2O2] and time shows that the decomposition of hydrogen peroxide is a first-order reaction.
The rate constant for a first-order reaction is equal to the negative of the slope of the plot of ln[H2O2] versus time where:
\[\ce{slope}=\dfrac{\textrm{change in }y}{\textrm{change in }x}=\dfrac{Δy}{Δx}=\dfrac{Δ\ln[\ce{H2O2}]}{Δt} \nonumber \]
To determine the slope of the line, we need two values of ln[H2O2] at different values of t (one near each end of the line is preferable). For example, the value of ln[H2O2] when t is 6.00 h is −0.693; the value when t = 12.00 h is −1.386:
\[\begin{align*}
\ce{slope}&=\mathrm{\dfrac{−1.386−(−0.693)}{12.00\: h−6.00\: h}}\\[4pt]
&=\mathrm{\dfrac{−0.693}{6.00\: h}}\\[4pt]
&=\mathrm{−1.155×10^{−2}\:h^{−1}}\\[4pt]
k&=\mathrm{−slope=−(−1.155×10^{−1}\:h^{−1})=1.155×10^{−1}\:h^{−1}}
\end{align*} \nonumber \]
Graph the following data to determine whether the reaction \(A⟶B+C\) is first order.
| Trial | Time (s) | [A] |
|---|---|---|
| 1 | 4.0 | 0.220 |
| 2 | 8.0 | 0.144 |
| 3 | 12.0 | 0.110 |
| 4 | 16.0 | 0.088 |
| 5 | 20.0 | 0.074 |
- Answer
-
The plot of ln[A] vs. t is not a straight line. The equation is not first order:
![A graph, labeled above as “l n [ A ] vs. Time” is shown. The x-axis is labeled, “Time ( s )” and the y-axis is labeled, “l n [ A ].” The x-axis shows markings at 5, 10, 15, 20, and 25 hours. The y-axis shows markings at negative 3, negative 2, negative 1, and 0. A slight curve is drawn connecting five points at coordinates of approximately (4, negative 1.5), (8, negative 2), (12, negative 2.2), (16, negative 2.4), and (20, negative 2.6).](https://chem.libretexts.org/@api/deki/files/121305/Figure_2.png?revision=1&size=bestfit&width=550&height=319)
Second-Order Integrated Rate Law
A reaction that is second-order overall can occur in two ways:
- One reactant with a reaction order of 2: rate = k[A]2
- Two reactants each with a reaction order of 1: rate = k[A][B]
In this section, we are focussing on the first case, which assesses whether the reaction order of a single reactant is second order. However, in the CHM135 laboratory we will explore how the reaction orders of multi-component rate laws can be determined.
The rate law for a second-order reaction is:
\[\ce{rate}=-\dfrac{\Delta [A]}{\Delta t}=k[A]^2 \nonumber \]
Changing deltas to differentials,
\[\ -\dfrac{d[A]}{dt} =k [A]^2 \nonumber \]
then rearranging the equation to separate the variables \([A]\) and \(t\),
\[\ \dfrac{1}{[A]^2} \: d[A]=-k \: dt \nonumber \]
we can integrate with respect to \([A]\) on the left from \([A]_o\) to \([A]_t\) and with respect to \(t\) on the right from 0 to t:
\[\int\limits_{[A]_o}^{[A]_t} \dfrac{1}{[A]^2}\, d[A] = \int\limits_{0}^{t} -k\, dt \nonumber\]
This gives us the second-order integrated rate law, which provides a linear expression for changes in reactant concentration as a function of time:
\[-(\dfrac{1}{[A]}-\dfrac{1}{[A]_0})=-kt \label{int2nd} \]
The integrated rate law for the second-order reaction can be rewritten in the form of a linear equation:
\[\begin{align*}
\dfrac{1}{[A]}&=kt+\dfrac{1}{[A]_0}\\[4pt]
y&=mx+b
\end{align*} \nonumber \]
For a second-order reaction, a plot of \(\dfrac{1}{[A]}\) versus \(t\) yields a straight line with a slope of \(k\) and an intercept of \(\dfrac{1}{[A]_0}\). If this plot is not a straight line, the reaction is not second order.
The reaction of butadiene gas (C4H6) with itself produces C8H12 gas as follows:
\[\ce{2C4H6}(g)⟶\ce{C8H12(g)} \nonumber \]
The reaction is second order with a rate constant equal to 5.76 × 10−2 L/mol/min under certain conditions. If the initial concentration of butadiene is 0.200 M, what is the concentration remaining after 10.0 min?
Solution
We use the integrated form of the rate law to answer questions regarding time. For a second-order reaction, we have:
\[\dfrac{1}{[A]}=kt+\dfrac{1}{[A]_0} \nonumber \]
\[\begin{align*}
\dfrac{1}{[A]}&=\mathrm{(5.76×10^{−2}\:L\: mol^{−1}\:min^{−1})(10\:min)+\dfrac{1}{0.200\:M}}\\[4pt]
\dfrac{1}{[A]}&=\mathrm{(5.76×10^{−1}\:M^{−1})+5.00\:M^{−1}}\\[4pt]
\dfrac{1}{[A]}&=\mathrm{5.58\:M^{−1}}\\[4pt]
[A]&=\mathrm{1.79×10^{−1}\:M}
\end{align*} \nonumber \]
Therefore 0.179 mol/L of butadiene remain at the end of 10.0 min, compared to the 0.200 mol/L that was originally present.
Using the reaction in an initial concentration of butadiene is 0.0200 M, what is the concentration remaining after 20.0 min?
- Answer
-
0.0195 mol/L
Test the data given to show whether the dimerization of C4H6 is a first- or a second-order reaction.
Solution
| Time (s) | [C4H6] (M) |
|---|---|
| 0 | 1.00 × 10−2 |
| 1600 | 5.04 × 10−3 |
| 3200 | 3.37 × 10−3 |
| 4800 | 2.53 × 10−3 |
| 6200 | 2.08 × 10−3 |
To distinguish a first-order reaction from a second-order reaction, we plot ln[C4H6] versus t and compare it with a plot of \(\mathrm{\dfrac{1}{[C_4H_6]}}\) versus t. The values needed for these plots follow.
| Time (s) | \(\dfrac{1}{[\ce{C4H6}]}\:(M^{−1})\) | ln[C4H6] |
|---|---|---|
| 0 | 100 | −4.605 |
| 1600 | 198 | −5.289 |
| 3200 | 296 | −5.692 |
| 4800 | 395 | −5.978 |
| 6200 | 481 | −6.175 |
As you can see, the plot of ln[C4H6] versus t is not linear, therefore the reaction is not first order. The plot of \(\dfrac{1}{[\ce{C4H6}]}\) versus t is linear, indicating that the reaction is second order.
![Two graphs are shown, each with the label “Time ( s )” on the x-axis. The graph on the left is labeled, “l n [ C subscript 4 H subscript 6 ],” on the y-axis. The graph on the right is labeled “1 divided by [ C subscript 4 H subscript 6 ],” on the y-axis. The x-axes for both graphs show markings at 3000 and 6000. The y-axis for the graph on the left shows markings at negative 6, negative 5, and negative 4. A decreasing slightly concave up curve is drawn through five points at coordinates that are (0, negative 4.605), (1600, negative 5.289), (3200, negative 5.692), (4800, negative 5.978), and (6200, negative 6.175). The y-axis for the graph on the right shows markings at 100, 300, and 500. An approximately linear increasing curve is drawn through five points at coordinates that are (0, 100), (1600, 198), (3200, 296), and (4800, 395), and (6200, 481).](https://chem.libretexts.org/@api/deki/files/121306/Figure_3.png?revision=1&size=bestfit&width=737&height=269)
Does the following data fit a second-order rate law?
| Trial | Time (s) | [A] (M) |
|---|---|---|
| 1 | 5 | 0.952 |
| 2 | 10 | 0.625 |
| 3 | 15 | 0.465 |
| 4 | 20 | 0.370 |
| 5 | 25 | 0.308 |
| 6 | 35 | 0.230 |
- Answer
-
Yes. The plot of \(\dfrac{1}{[A]}\) vs. t is linear:
![A graph, with the title “1 divided by [ A ] vs. Time” is shown, with the label, “Time ( s ),” on the x-axis. The label “1 divided by [ A ]” appears left of the y-axis. The x-axis shows markings beginning at zero and continuing at intervals of 10 up to and including 40. The y-axis on the left shows markings beginning at 0 and increasing by intervals of 1 up to and including 5. A line with an increasing trend is drawn through six points at approximately (4, 1), (10, 1.5), (15, 2.2), (20, 2.8), (26, 3.4), and (36, 4.4).](https://chem.libretexts.org/@api/deki/files/121307/Figure_4.png?revision=1&size=bestfit&width=417&height=281)
The Half-Life of a Reaction
The half-life of a reaction (t1/2) is the time required for the reactant concentration to decrease by half. In each succeeding half-life, half of the remaining concentration of the reactant is consumed. For a first-order reaction, the half-life remains constant and does not depend on the starting concentration.
For example, the decomposition of hydrogen peroxide (H2O2) follows first-order kinetics. Figure \(\PageIndex{4}\) shows its concentration decreasing over time in a series of four half-lives:
- First half-life: [H2O2] decreases from 1.00 M to 0.500 M
- Second half-life: [H2O2] decreases from 0.500 M to 0.250 M
- Third half-life: [H2O2] decreases from 0.250 M to 0.125 M
- Fourth half-life: [H2O2] decreases from 0.125 M to 0.0625 M
From this plot, we can see that each half-life is 6 hours, confirming that the half-life of a first-order reaction remains constant over time.

Figure \(\PageIndex{4}\): The decomposition of H2O2 is a first-order reaction. At a particular temperature, the half-life of the reaction is 6 hours, meaning that half of the H2O2 decomposes every 6 hours. This plot shows 4 successive half-lives where the [H2O2] decreases from 1 M to 0.5 M in the first, 0.500 M to 0.250 M in the second, 0.250 M to 0.125 M in the third and 0.125 M to 0.0625 M in the fourth.
The half-life of a first-order reaction can be derived from its integrated rate law:
\[\ln\dfrac{[A]_0}{[A]_t}=kt \nonumber \]
Solving for t:
\[t=\dfrac{ln\dfrac{[A]_0}{[A]_t}}{k}\nonumber \]
Since half-life is the time when [A] is half of its initial value, we substitute [A]t = 1/2[A]0 and set t = t1/2:
\[t_{1/2}=\dfrac{ln\dfrac{[A]_0}{\dfrac{1}{2}[A]_0}}{k}\nonumber \]
Simplifying:
\[t_{1/2}=\dfrac{\ln2}{k}=\dfrac{0.693}{k}\nonumber \]
This equation shows that the half-life of a first-order reaction is independent of reactant concentration and depends only on the rate constant k. We can also see that the half-life of a first-order reaction is inversely proportional to the rate constant k. A fast reaction (larger k) will have a shorter half-life; a slow reaction (smaller k) will have a longer half-life. This relationship makes half-life calculations particularly useful in fields like pharmacology (drug metabolism) and nuclear chemistry (radioactive decay), where processes are often first-order and knowing how long a substance persists is essential.
Calculate the rate constant for the first-order decomposition of hydrogen peroxide in water at 40 °C, using the data given below:
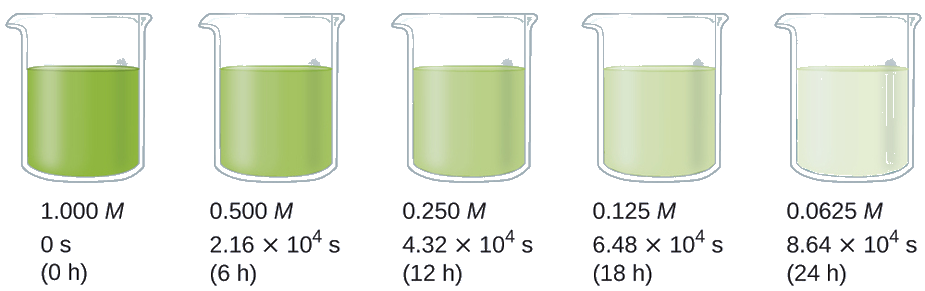
Solution
The half-life for the decomposition of H2O2 is 2.16 × 104 s:
\[\begin{align*}
t_{1/2}&=\dfrac{0.693}{k}\\[4pt]
k&=\dfrac{0.693}{t_{1/2}}=\dfrac{0.693}{2.16×10^4\:\ce s}=3.21×10^{−5}\:\ce s^{−1}
\end{align*} \nonumber \]
The first-order radioactive decay of iodine-131 exhibits a rate constant of 0.138 d−1. What is the half-life for this decay?
- Answer
-
5.02 d.
The half-life (t1/2) of a zero-order reaction can be determined using the zero-order integrated rate law:
\[[A]=−kt+[A]_0 \nonumber \]
Substituting t = t1/2 and [A]t = 1/2[A]0:
\[\begin{align*}
\dfrac{[A]_0}{2}&=−kt_{1/2}+[A]_0\\[4pt]
kt_{1/2}&=\dfrac{[A]_0}{2}\\[4pt]
t_{1/2}=\dfrac{[A]_0}{2k}
\end{align*} \nonumber \]
This equation demonstrates that the half-life of a zero-order reaction depends on the initial concentration ([A]0) and that as the reaction proceeds, the half-life decreases. This might seem strange, if the half-life is decreases... does that mean the rate of the reaction is increasing as time going on? No, that is (obviously) possible. In a zero-order reaction the rate of the reaction is unchanged with reactant concentration and so as the reaction proceeds the half-life gets shorter because there is simply less material present and so it will not take as long to half that smaller amount as the rate is not changing.
The half-life (t1/2) of a second-order reaction can be determined using the second-order integrated rate law:
\[\dfrac{1}{[A]}=kt+\dfrac{1}{[A]_0} \nonumber \]
Substituting t = t1/2 and [A]t = 1/2[A]0:
\(\begin{align*}
\dfrac{1}{[A]}−\dfrac{1}{[A]_0}&=kt\\[4pt]
\dfrac{1}{\dfrac{1}{2}[A]_0}−\dfrac{1}{[A]_0}&=kt_{1/2}\\[4pt]
\dfrac{2}{[A]_0}−\dfrac{1}{[A]_0}&=kt_{1/2}\\[4pt]
\dfrac{1}{[A]_0}&=kt_{1/2}\\[4pt]
t_{1/2}&=\dfrac{1}{k[A]_0}
\end{align*}\nonumber \)
This equation shows that the half-life of a second-order reaction is inversely proportional to the initial concentration ([A]0) and so as the reaction proceeds, the half-life increases. This increase in half-life means the reaction is proceeding at a slower rate as the reactant is consumed.
Comparing Zero-, First-, and Second-Order Reactions
The following table summarizes the key differences between zero-, first-, and second-order reactions:
| Zero-Order | First-Order | Second-Order | |
|---|---|---|---|
| Rate law | rate = k | rate = k[A] | rate = k[A]2 |
| Units of k | M s−1 | s−1 | M−1 s−1 |
| Integrated rate law | [A]t = −kt + [A]0 | ln[A]t = −kt + ln[A]0 | \(\dfrac{1}{[A]_t}=kt+\left(\dfrac{1}{[A]_0}\right)\) |
| Graph for linear fit | [A] vs. t | ln[A] vs. t | \(\dfrac{1}{[A]}\) vs. t |
| Slope in graph | k = −slope | k = −slope | k = +slope |
| Half-life | \(t_{1/2}=\dfrac{[A]_0}{2k}\) | \(t_{1/2}=\dfrac{0.693}{k}\) | \(t_{1/2}=\dfrac{1}{[A]_0k}\) |
Summary
Rate laws describe how the rate of a reaction depends on reactant concentration. Reaction orders and the rate constant within the rate law can be determined using the method of initial rates or graphical analysis of concentration vs. time data.
Integrated rate laws describe how reactant concentrations change over time and are derived by integrating the rate laws.
The half-life of a reaction is the time required to decrease the amount of a given reactant by one-half. In a zero-order reaction, the half-life decreases as the reaction proceeds. In a first-order reaction, the half-life is constant and does not depend on concentration. In a second-order reaction, the half-life increases as the reaction proceeds.