
2.9: Organic Acids and Organic Bases
- Page ID
- 31392
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)After completing this section, you should be able to
- predict the relative acidity of two organic molecules from their structures.
- predict the relative basicity of two organic molecules from their structures.
This page explains the acidity of simple organic acids and looks at the factors which affect their relative strengths.
Organic acids as weak acids
For the purposes of this topic, we are going to take the definition of an acid as "a substance which donates hydrogen ions (protons) to other things". We are going to get a measure of this by looking at how easily the acids release hydrogen ions to water molecules when they are in solution in water.
An acid in solution sets up this equilibrium:
\[AH_{(aq)} + H_2O_{(l)} \rightleftharpoons A^-_{(aq)} + H_3O^+_{(aq)} \nonumber \]
A hydronium ion is formed together with the anion (negative ion) from the acid. This equilibrium is sometimes simplified by leaving out the water to emphasize the ionization of the acid.
\[AH_{(aq)} \rightleftharpoons A^-_{(aq)} + H^+_{(aq)} \nonumber \]
If you write it like this, you must include the state symbols - "(aq)". Writing H+(aq) implies that the hydrogen ion is attached to a water molecule as H3O+. Hydrogen ions are always attached to something during chemical reactions.
The organic acids are weak in the sense that this ionization is very incomplete. At any one time, most of the acid will be present in the solution as un-ionized molecules. For example, in the case of dilute ethanoic acid, the solution contains about 99% of ethanoic acid molecules - at any instant, only about 1% have actually ionized. The position of equilibrium therefore lies well to the left.
Weak acid polarization
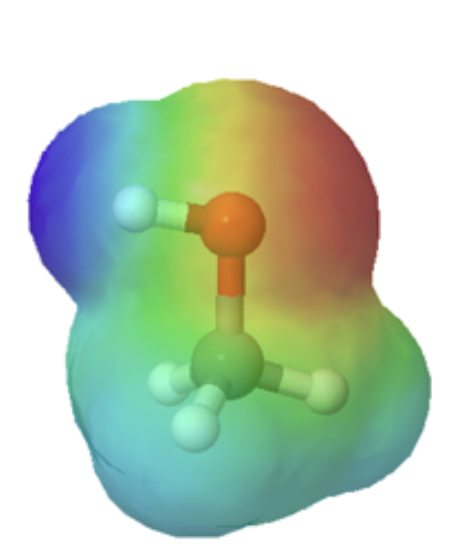
Organic acids can usually can be characterized in electrostatic potential maps by the presence of of a positively polarized hydrogen atom shown in blue. When looking at the maps below, methanol has a slightly polarized O-H bond and is considered very weakly acidic. The O-H bond in methyl amine is less polarized, as shown by the lighter blue color around the hydrogen, making it less acidic than methanol. However, the C-H bond in ethane lack virtually any polarity, as shown by the lack of a blue color, making it non-acidic. The following discussion will explain the difference in acidity of these and other organic molecules molecules.
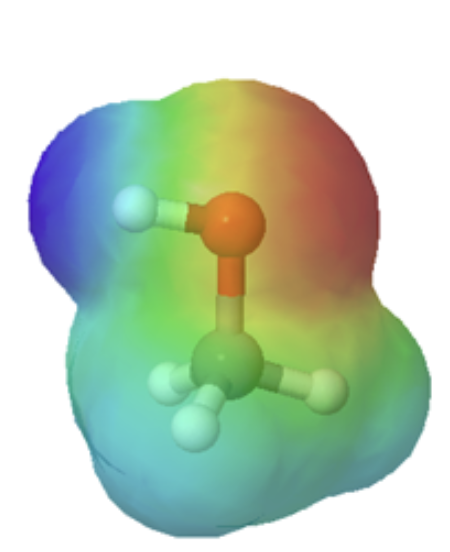 methanol |
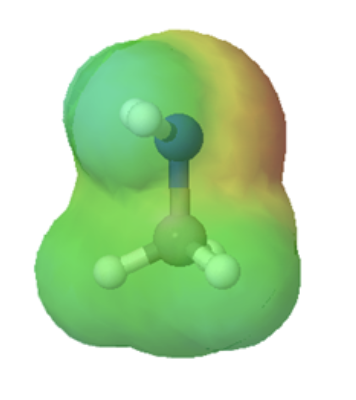 methylamine |
 ethane |
Comparing the strengths of weak acids
Acid strength is strongly correlated to stability of the conjugate base that will form by removing a proton. In order to analyze how acidic a molecule is likely to be, then you need to estimate the stability of its conjugate base.
Stabilization of the Conjugate Base - Four Main Considerations:
- Size and electronegativity of the atom holding the charge
- Can the charge be delocalized by resonance?
- Are there any inductive effects?
- Hybridization of orbital holding the charge
These considerations are listed in order of importance and are explained individually, but must be looked at collectively.
-
Size and Electronegativity Effects in Acidity
When comparing elements, it depends on the positional relationship of the elements on the periodic table. When moving a period (aka across a row) of the main group elements, the valence electrons all occupy orbitals in the same shell. These electrons have comparable energy, so this factor does not help us discern differences relative stability. Differences in electronegativity are now the dominant factor. This trend is shown when comparing the pKa values of ethane, methyl amine, and methanol which reflects the relative electronegativities of the C < N < O. The key to understanding this trend is to consider the hypothetical conjugate base in each case: the more stable the conjugate base, the stronger the acid. In general, the more electronegative an atom, the better it is able to bear a negative charge. In the ethyl anion, the negative charge is borne by carbon, in the methylamine anion by nitrogen, and in the methoxide anion by an oxygen. Remember the periodic trend in electronegativity: it also increases as we move from left to right along a row, meaning that oxygen is the most electronegative of three elements being considered. This makes the negative charge on the methoxide anion the most stable of the three conjugate bases and methanol the strongest of the three acids. Likewise, carbon is the least electronegative making ethane the weakest of the three acids.



Within a Group (aka down a column) As we move down the periodic table, the electrons are occupying higher energy subshells creating a larger atomic size and volume. As the volume of an element increases, any negative charge present tends to become more spread out which decreases electron density and increases stability. The figure below shows spheres representing the atoms of the s and p blocks from the periodic table to scale, showing the two trends for the atomic radius.
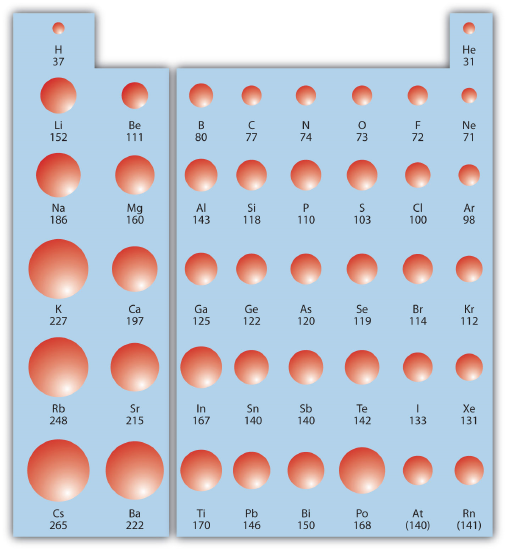
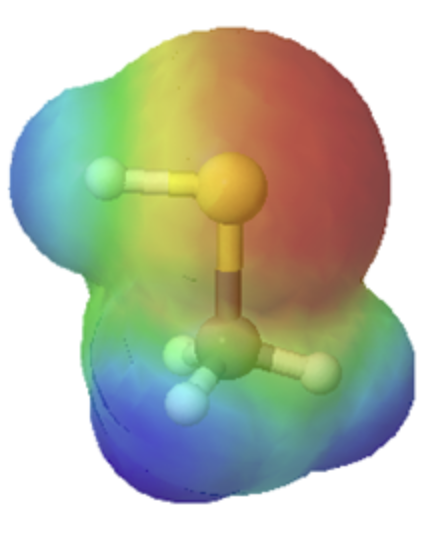
This relationship of atomic size and electron density is illustrated when we compare the relative acidities of methanol, CH3OH, with methanethiol, CH3SH. The lower pKa value of 10.4 for methanethiol indicates that it is a stronger acid than methanol with a pKa value of 15.5. It is important to remember that neither compound is considered an acid. These relationships become useful when trying to deprotonate compounds to increase their chemical reactivity in non-aqueous reaction conditions.


The difference in size can easily be seen when looking at the electrostatic potential maps for methanol (Left) and methanethiol (Right). The sulfur atom methanethiol is larger than the oxygen atom in methanol. The larger size of sulfur will be better able to delocalize and stabilize the negative charge in its conjugate base metanethiolate.
-
Resonance Effects in Acidity
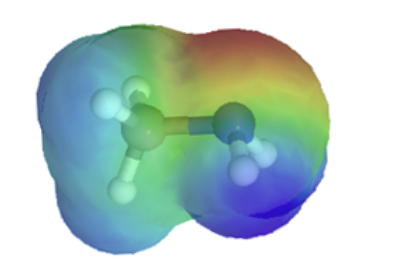
This section will focus on how the resonance structures of different organic groups contributes to their relative acidity even though the same element acts as the proton donor. When evaluating conjugate bases for the presence of resonance contributors, remember to look for movable electrons (lone pairs and pi bonding electrons). Delocalizing electrons over two or more atoms spreads out the electron density, increasing the stability of the conjugate base, and increasing the acidity of the corresponding acid. A classic example compares the relative acidity of ethanol and acetic acid, but the conclusions we reach will be equally valid for all alcohol and carboxylic acid groups. Despite the fact that they are both oxygen acids, the pKa values of ethanol and acetic acid are very different.


In both species, the negative charge on the conjugate base is held by an oxygen, so periodic trends cannot be invoked. For acetic acid, however, there is a key difference: a resonance contributor can be drawn in which the negative charge is drawn on the second oxygen of the group. The two resonance forms for the conjugate base are equal in energy, according to our ‘rules of resonance’ (Section 2.5). What this means is that the negative charge on the acetate ion is not located on one oxygen or the other: rather it is shared between the two. Chemists use the term ‘delocalization of charge’ to describe this situation. In the ethoxide ion, by contrast, the negative charge is ‘locked’ on the single oxygen. This stabilization leads to a markedly increased acidity.
The delocalization of charge by resonance has a very powerful effect on the reactivity of organic molecules, enough to account for the difference of nearly 12 pKa units between ethanol and acetic acid (and remember, pKa is a log expression, so we are talking about a difference of over 1012 between the acidity constants for the two molecules). The acetate ion is much more stable than the ethoxide ion, due to the effects of resonance delocalization.
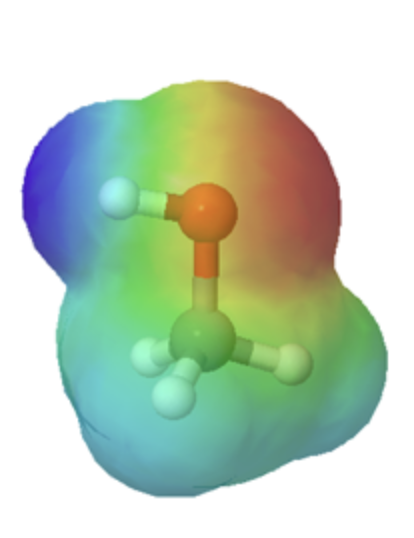
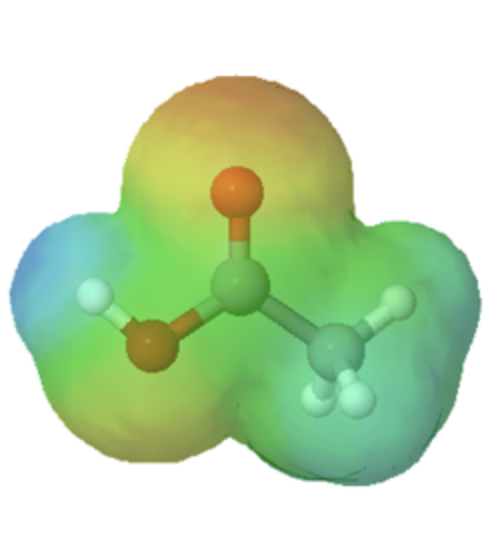
The effects of conjugation can be seen when comparing the electrostatic potential maps of ethanol and acetic acid. Conjugation creates a greater polarization in the O-H bond in acetic acid as shown by its darker blue color.
|
ethanol |
acetic acid |


Why is Phenol Acidic?
Compounds like alcohols and phenol which contain an -OH group attached to a hydrocarbon are very weak acids. Alcohols are so weakly acidic that, for normal lab purposes, their acidity can be virtually ignored. However, phenol is sufficiently acidic for it to have recognizably acidic properties - even if it is still a very weak acid. A hydrogen ion can break away from the -OH group and transfer to a base. For example, in aqueous solution:


Since phenol is a very weak acid, the position of equilibrium lies well to the left. However, phenol can lose a hydrogen ion because the phenoxide ion (or phenolate ion - the two terms can be used interchangeably) formed is stabilized due to resonance. The negative charge on the oxygen atom is delocalized around the ring since one of the lone pairs on the oxygen atom can be in a p orbital and overlap with the pi electrons on the benzene ring.

This overlap leads to a delocalization which extends from the ring out over the oxygen atom. As a result, the negative charge is no longer entirely localized on the oxygen, but is spread out around the whole ion. Spreading the charge around makes the ion more stable than it would be if all the charge remained on the oxygen. However, oxygen is the most electronegative element in the ion and the delocalized electrons will be drawn towards it. That means that there will still be a lot of charge around the oxygen which will tend to attract the hydrogen ion back again. That is why phenol is only a very weak acid.
This explains why phenol is a much stronger acid than cyclohexanol. As can be seen in the following energy diagram, resonance stabilization is increased for the conjugate base of phenol vs. cyclohexanol after removal of a proton.

The resonance stabilization in these two cases is very different. An important principle of resonance is that charge separation diminishes the importance of contributors to the resonance hybrid. The contributing structures to the phenol hybrid all suffer charge separation, resulting in very modest stabilization of this compound. On the other hand, the phenolate anion is already charged, and the canonical contributors act to disperse the charge, resulting in a substantial stabilization of this species. The conjugate bases of simple alcohols are not stabilized by charge delocalization, so the acidity of these compounds is similar to that of water. An energy diagram showing the effect of resonance on cyclohexanol and phenol acidities is shown on the right. Since the resonance stabilization of the phenolate conjugate base is much greater than the stabilization of phenol itself, the acidity of phenol relative to cyclohexanol is increased. Supporting evidence that the phenolate negative charge is delocalized on the ortho and para carbons of the benzene ring comes from the influence of electron-withdrawing substituents at those sites.
Acidity of hydrogen α (alpha) to carbonyl
Alkyl hydrogen atoms bonded to a carbon atom in a α (alpha) position (directly adjacent) relative to a C=O group display unusual acidity. While the pKa values for alkyl C-H bonds in is typically on the order of 40-50, pKa values for these alpha hydrogens is more on the order of 19-20. This is almost exclusively due to the resonance stabilization of the product carbanion, called an enolate, as illustrated in the diagram below. The effect of the the stabilizing C=O is seen when comparing the pKa for the α hydrogens of aldehydes (~16-18), ketones (~19-21), and esters (~23-25).

-
Inductive Effects
The inductive effect is an experimentally observed effect of the transmission of charge through a chain of atoms in a molecule, resulting in a permanent dipole in a bond. For example, in a carboxylic acid group the presence of chlorine on adjacent carbons increases the acidity of the carboxylic acid group. A chlorine atom is more electronegative than hydrogen, and thus is able to ‘induce’, or ‘pull’ electron density towards itself, away from the carboxylate group. This further spreads out the electron density of the conjugate base, which has a stabilizing effect. In this context, the chlorine substituent is called an electron-withdrawing group. Notice that the pKa-lowering effect of each chlorine atom, while significant, is not as dramatic as the delocalizing resonance effect illustrated by the difference in pKa values between an alcohol and a carboxylic acid. In general, resonance effects are more powerful than inductive effects.

negative charge is delocalized by being pulled out onto chlorine atom
Compare the pKa values of acetic acid and its mono-, di-, and tri-chlorinated derivatives:
%252C_chloroacetic_acid_(2.8)%252C_dichloroacetic_acid_(1.3)%252C_and_trichloroacetic_acid_(0.64).svg?revision=1&size=bestfit&width=506&height=92)
The inductive effects of chlorine can be clearly seen when looking at the electrostatic potential maps of acetic acid (Left) and trichloroacetic acid (Right). The O-H bond in trichloroacetic acid is highly polarized as shown by the dark blue color. This illustrates that tricholoracetic acid is a much stronger acid than acetic acid.
|
|
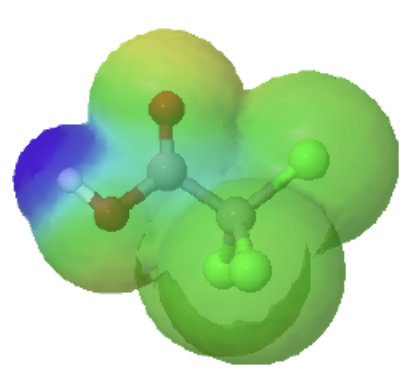  trichloroacetic acid |

Because the inductive effect depends on electronegativity, fluorine substituents have a more pronounced pKa-lowering effect than chlorine substituents.
_and_trifluoroacetic_acid_(pka_-0.25).svg?revision=1&size=bestfit&width=385&height=131)
In addition, the inductive takes place through covalent bonds, and its influence decreases markedly with distance – thus a chlorine two carbons away from a carboxylic acid group has a decreased effect compared to a chlorine just one carbon away. 2-chloropropanoic acid has a pKa of 2.8 while for 3-chloropropanoic acid, the pKa is 4.0.
_and_3-chloropropanoic_acid_(pKa_4.0).svg?revision=1&size=bestfit&width=383&height=142)
Alkyl groups (hydrocarbons) are weak inductive electron donators. In this case the inductive effect pushes electron density onto the conjugate base, causing the electron density to become more concentrated and producing a destabilizing effect.

more negative charge pushed towards already negative end
The inductive effects of alkyl groups causes a significant variation in the acidities of different carboxylic acids. Notice that the inductive effect drops off after the alkyl chain is about three carbons long.
| pKa | |
| HCOOH (Methanoic Acid or Formic Acid) | 3.75 |
| CH3COOH (Ethanoic Acid or Acetic Acid) | 4.76 |
| CH3CH2COOH (Propanoic Acid) | 4.87 |
| CH3CH2CH2COOH (Butanoic Acid) | 4.82 |
-
Orbital Hybridization
The hybridization of an orbital affects its electronegativity. Within a shell, the s orbitals occupy the region closer to the nucleus than the p orbitals. Therefore, the spherical s orbitals are more electronegative than the lobed p orbitals. The relative electronegativity of hybridized orbitals is sp > sp2 > sp3 since the percentage of s-character is decreasing as more p-orbitals are added to the hybrids. This trend indicates the sp hybridized orbitals are more stable with a negative charge than sp3 hybridized orbitals. The table below shows how orbital hybridization influences relative acidity.
| compound | conjugate base | hybridization | s character | pKa | |
 |
 |
sp3 | 25% | 50 | weakest acid |
 |
 |
sp2 | 33% | 44 | ↓ |
 |
 |
36 | ↓ | ||
 |
 |
sp | 50% | 25 | ↓ |
 |
.svg?revision=1) |
16 | strongest acid |
Comparing the Strengths of Weak Bases
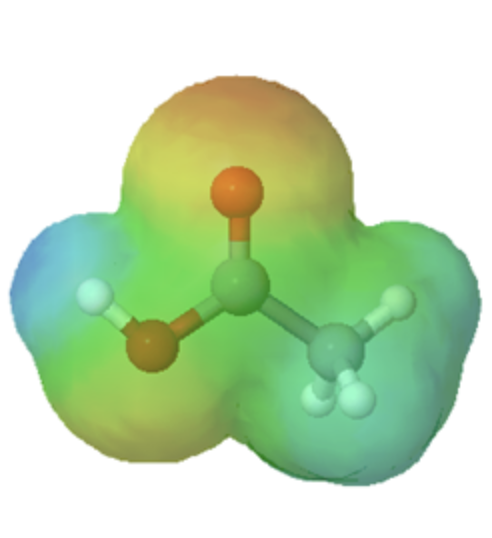
Technically, organic bases are characterized by the presence of an atom with lone pair electrons. These lone pairs contain a high electron density, which is shown red in the electrostatic potential maps, and can bond to H+. Below are the maps of methanol, methyl amine, and acetone. All three compounds can be protonated with a sufficiently strong acid. Note, that all three of these compounds also have the ability to donate a proton when reacted with a strong enough base. Whether these compounds act as a acid or base depends on the conditions.
  methylamine |
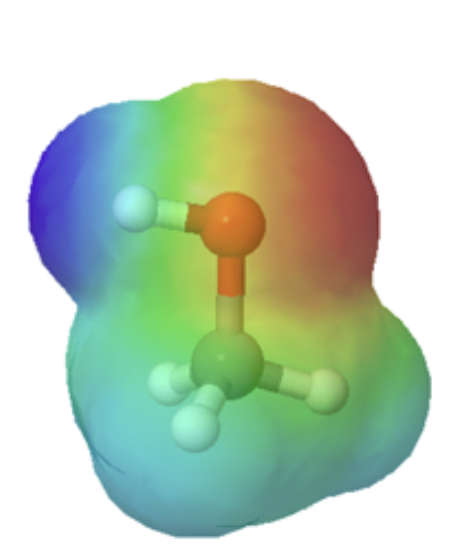  methanol |
  acetone |
It is common to compare basicity's quantitatively by using the pKa's of their conjugate acids rather than their pKb's. Since pKa + pKb = 14, the higher the pKa the stronger the base, in contrast to the usual inverse relationship of pKa with acidity. Recall that ammonia (NH3) acts as a base because the nitrogen atom has a lone pair of electrons that can accept a proton. The conjugate acid of most simple alkyl amines have pKa's in the range 9.5 to 11.0, and their water solutions are basic (have a pH of 11 to 12, depending on concentration). This can be illustrated by the reaction below where an amine removes a proton from water to form substituted ammonium (e.g. NH4+) ions and hydroxide (OH−) ions:

Amines are one of the only neutral functional groups which are considered basic. This is a direct consequence of the presence of the unshared electron pair on the nitrogen. The unshared electron pair is less tightly held by the nitrogen of an amine than the corresponding oxygen of an alcohol, which makes it more available to act as a base. As a specific example, methylamine reacts with water to form the methylammonium ion and the OH− ion.

All of the have similarities to ammonia and so we'll start by looking at the reason for its basic properties. For the purposes of this topic, we are going to take the definition of a base as "a substance which combines with hydrogen ions (protons)". We are going to get a measure of this by looking at how easily the bases take hydrogen ions from water molecules when they are in solution in water.
Ammonia in solution sets up this equilibrium:
\[ NH_3 + H_2O \rightleftharpoons NH_4^+ + OH^-\tag{2.10.1} \]
An ammonium ion is formed together with hydroxide ions. Because the ammonia is only a weak base, it doesn't hang on to the extra hydrogen ion very effectively and so the reaction is reversible. At any one time, about 99% of the ammonia is present as unreacted molecules. The position of equilibrium lies well to the left.
The ammonia reacts as a base because of the active lone pair on the nitrogen. Nitrogen is more electronegative than hydrogen and so attracts the bonding electrons in the ammonia molecule towards itself. That means that in addition to the lone pair, there is a build-up of negative charge around the nitrogen atom. That combination of extra negativity and active lone pair attracts the new hydrogen from the water.

When looking at the table below, it is clear that the basicity of nitrogen containing compounds is greatly influenced by their structures. The variance in the basicity of these compounds can mostly be explained by the effects of electron delocalization discussed above.
Table \(\PageIndex{1}\): pKa of conjugate acids of a series of amines.
| Compound |  |
 |
 |
 |
 |
 |
 |
 |
 |
 |
|---|---|---|---|---|---|---|---|---|---|---|
| pKa | 11.0 | 10.7 | 10.7 | 9.3 | 5.2 | 4.6 | 1.0 | 0.0 | -1.0 | -10.0 |
Inductive Effects in Nitrogen Basicity
Alkylamines are more basic than ammonia since alkyl groups donate electrons to the more electronegative nitrogen. This inductive effect makes the electron density on the alkylamine nitrogen greater than the nitrogen of ammonium. That means that there will be a small amount of extra negative charge built up on the nitrogen atom. That extra negativity around the nitrogen makes the lone pair even more attractive towards hydrogen ions. Correspondingly, primary, secondary, and tertiary alkyl amines are more basic than ammonia.

methyl group pushes electron density toward the nitrogen, making it more basic
Making the nitrogen more negative helps the lone pair to pick up a hydrogen ion. What about the effect on the positive methylammonium ion formed? Is this more stable than a simple ammonium ion? Compare the methylammonium ion with an ammonium ion:

In the methylammonium ion, the positive charge is spread around the ion by the "electron-pushing" effect of the methyl group. The more you can spread charge around, the more stable an ion becomes. In the ammonium ion there is not any way of spreading the charge.
To summarize:
- The nitrogen is more negative in methylamine than in ammonia, and so it picks up a hydrogen ion more readily.
- The ion formed from methylamine is more stable than the one formed from ammonia, and so is less likely to shed the hydrogen ion again.
Taken together, these mean that methylamine is a stronger base than ammonia.
| Compound | pKa |
| NH3 | 9.3 |
| CH3NH2 | 10.66 |
| (CH3)2NH | 10.74 |
| (CH3)3N | 9.81 |
Resonance Effects in Nitrogen Basicity
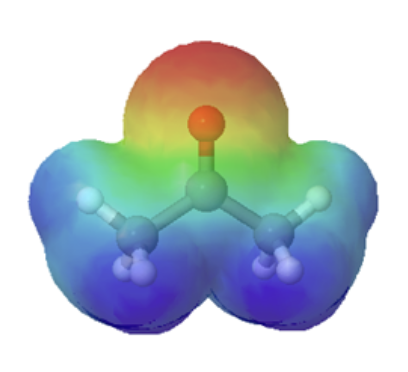
The resonance effect also explains why a nitrogen atom is basic when it is in an amine, but not significantly basic when it is part of an amide group. While the lone pair of electrons in an amine nitrogen is localized in one place, the lone pair on an amide nitrogen is delocalized by resonance. The lone pair is stabilized by resonance delocalization. Here’s another way to think about it: the lone pair on an amide nitrogen is not available for bonding with a proton – these two electrons are too stable being part of the delocalized pi-bonding system. The electrostatic potential map show the effect of resonance on the basicity of an amide. The map shows that the electron density, shown in red, is almost completely shifted towards the oxygen. This greatly decreases the basicity of the lone pair electrons on the nitrogen in an amide.
  amine pKa of conjugate acid ~ 11 |
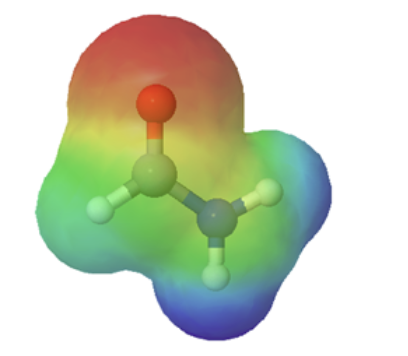  amide pKa of conjugate acid ~ −1 |
Aniline, the amine analog of phenol, is substantially less basic than an amine (as evidenced by the pKa of the conjugate acids).
|
|
aniline pKa of conjugate acid ~ 5 |
We can use the same reasoning that we used when comparing the acidity of a phenol to that of an alcohol. In aniline, the lone pair on the nitrogen atom is stabilized by resonance with the aromatic pi system, making it less available for bonding and thus less basic.

lone pair is stabilized through resonance
In these cases, you seem to be breaking the same oxygen-hydrogen bond each time, and so you might expect the strengths to be similar. The most important factor in determining the relative acid strengths of these molecules is the nature of the ions formed. You always get a hydronium ion - so that's constant - but the nature of the anion (the negative ion) varies markedly from case to case.
Exercises
Select the more basic from each of the following pairs of compounds.
(a)
(b)
- Answer
-
a)
The lone pair of electrons on the amide nitrogen are less available to react with a proton.
(b) NaOH -- The hydroxide has a negative charge with three lone pairs of electrons that can react with a proton.
The 4-methylbenzylammonium ion has a pKa of 9.51, and the butylammonium ion has a pKa of 10.59. Which is more basic? What's the pKb for each compound?
- Answer
-
The butylammonium is more basic. Remember that pKa+pKb = 14. The pKb for butylammonium is 3.41, the pKb for 4-methylbenzylammonium is 4.49.