
1.6: The Imperfect Solid State
- Page ID
- 408529

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)INTRODUCTION
Real crystals are never perfect: they always contain a considerable density of defects and imperfections that affect their physical, chemical, mechanical and electronic properties. The existence of defects also plays an important role in various technological processes and phenomena such as annealing, precipitation, diffusion, sintering, oxidation and others. It should be noted that defects do not necessarily have adverse effects on the properties of materials. There are many situations in which a judicious control of the types and amounts of imperfections can bring about specific characteristics desired in a system. This can be achieved by proper processing techniques. In fact, “defect engineering” is emerging as an important activity.
All defects and imperfections can be conveniently considered under four main divisions: point defects, line defects or dislocations, planar defects or interfacial or grain boundary defects, and volume defects. We can also add here macroscopic or bulk defects such as pores, cracks and foreign inclusions that are introduced during production and processing of the solid state. Point defects are inherent to the equilibrium state and thus determined by temperature, pressure and composition of a given system. The presence and concentration of other defects, however, depend on the way the solid was originally formed and subsequently processed.
Briefly consider the effects of imperfections or crystal defects on a few important properties of solids. The electrical behavior of semiconductors, for example, is largely controlled by crystal imperfections. The conductivity of silicon can thus be altered in type (n or p) and by over eight orders of magnitude through the addition of minute amounts of electrically active dopant elements. In this case, each atom of dopant, substitutionally incorporated, represents a point defect in the silicon lattice. The fact that such small amounts of impurity atoms can significantly alter the electrical properties of semiconductors is responsible for the development of the transistor and has opened up the entire field of solid state device technology. Practically none of the semiconducting properties that led to these engineering accomplishments are found in a “perfect” crystal. They are properties peculiar to the defective solid state.
The existence of dislocations (line defects) in crystals provides a mechanism by which permanent change of shape or mechanical deformation can occur. A crystalline solid free of dislocations is brittle and practically useless as an engineering material. While the existence of dislocations in crystals insures ductility (ability to deform), the theoretical strength of crystalline solids is drastically reduced by their presence.
We should recognize that dislocations play a central role in the determination of such important properties as strength and ductility. In fact, virtually all mechanical properties of crystalline solids are to a significant extent controlled by the behavior of line imperfections.
The ability of a ferromagnetic material (such as iron, nickel or iron oxide) to be magnetized and demagnetized depends in large part on the presence of two-dimensional imperfections known as Bloch walls. These interfaces are boundaries between two regions of the crystal which have a different magnetic state. As magnetization occurs, these defects migrate and by their motion provide the material with a net magnetic moment. Without the existence of Bloch walls all ferromagnetic materials would be permanent magnets. In fact, electromagnets would not exist if it were not for this type of defect.
The presence of surface defects such as cracks causes brittle materials like glass to break at small applied stresses. This fact is familiar to anyone who has broken a glass tube by first filing a small notch (or crack) into the surface. Removal of cracks from the surface of glass either by etching in hydrofluoric acid or by flame polishing almost always raises the fracture strength. For example, glass in the absence of any surface cracks has a fracture strength of \(\sim 10^{10} \mathrm{Newton} / \mathrm{m}^2\) (as opposed to real glass which has a fracture strength of \(\sim 0^7\) Newton \(/ \mathrm{m}^2\) ).
POINT DEFECTS
Formation of Point Defects
An incontrovertible law of nature states: “Nothing is perfect”. This law applies to humans as well as to the inorganic world of crystalline solids and can be formulated as the 2nd law of thermodynamics:
\[\mathrm{F}=\mathrm{H}-\mathrm{TS} \tag{1}\]
where \(\mathrm{F}\) is the free energy of a given system, \(\mathrm{H}\) is the heat content or enthalpy and TS is the entropy, or disorder, term. If a reaction takes place at a temperature \(T\), we find the change in \(\mathrm{F}(\Delta \mathrm{F})\) related to a change in \(\mathrm{H}(\Delta \mathrm{H})\), the heat content, and possibly also a change in TS \((T \Delta S)\). Such is the case when defects are formed in a perfect solid: The energy distribution in a solid (Maxwell-Boltzmann) suggests that a number of individual atoms may acquire enough thermal energy to be displaced from the equilibrium lattice site into an interstitial position. This process of point defect formation requires energy and leads to lattice strain which constitutes, as discussed earlier, an increase in the heat content of the system (\(\Delta H\) is positive and increases linearly with the number of defects formed). The departure from perfection by the generation of defects leads to disorder (\(\Delta S\) is positive). The magnitude of disorder generated (\(\Delta S\)) is very large during the initial step from perfection to slight disarray, but the increase in disorder (with a given number of defects generated) decreases as the overall disorder increases.
Correspondingly the term \(T \Delta S\) drops rapidly at the beginning and then flattens out. The net result (fig. 1), free energy, exhibits a minimum for a certain number of defects in the
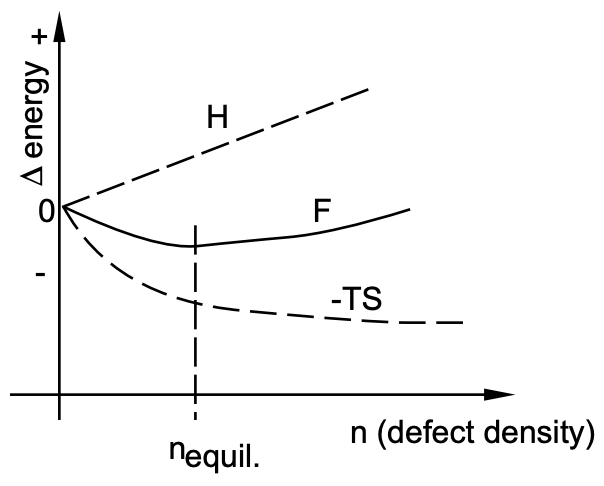
solid [equilibrium defect density = f (temperature)]; the \(F_{\text{minimum}}\) suggests also that the transition from perfection to equilibrium defect structure is spontaneous: it occurs naturally!
While the detailed mechanisms for the formation of atomic vacancies in solids are still the subject of extensive research, the associated equilibrium energetics are clear: calculations of the thermal energy of atoms in a lattice show that the average vibrational energy of lattice atoms is much less than \(1 \mathrm{eV}\) (the approximate energy change associated with vacancy formation, i.e., the least amount of energy required to form a vacancy) at room temperature. Therefore a lattice atom will only acquire the energy \(\Delta \mathrm{H}_{\mathrm{d}}\), the energy required to form the defect, upon the occurrence of a large energy fluctuation. Since the relative probability of an atom having an energy \(\Delta \mathrm{H}_d\) or more in excess of the ground state energy is \(e^{-\Delta H_d / k T}\), the probability that an atomic site is vacant varies in the same way. In a (molar) crystal containing \(\mathrm{N}\) atomic sites, the number \(\mathrm{n}_{\mathrm{d}}\) of vacant sites is, therefore,
\[\mathrm{n}_{\mathrm{d}}=\mathrm{ANe}^{-\Delta \mathrm{H}_{\mathrm{d}} / \mathrm{kT}} \tag{2}\]
where
- \(\mathrm{n}_{\mathrm{d}}\) is the number of defects (in equilibrium at \(\mathrm{T}\) )
- \(\mathrm{N}\) is the total number of atomic sites per mole
- \(\Delta \mathrm{H}_{\mathrm{d}}\) is the energy necessary to form the defect
- \(\mathrm{T}\) is the absolute temperature \((\mathrm{K})\)
- \(\mathrm{k}\) is the Boltzmann constant
- \(\mathrm{A}\) is a proportionality constant
Point Defects in “Pure” Metallic Systems
Point defects in “pure” crystalline metals are defects of atomic dimensions, such as impurity atoms, the absence of a matrix atom and/or the presence of a matrix atom in the wrong place. Some of these point defects are shown in fig. 2. An impurity atom that occupies a normal lattice site is called a substitutional impurity atom and an impurity atom found in the interstice between matrix atoms is called an interstitial impurity atom. Whether a foreign atom will occupy a substitutional

or interstitial site depends largely on the size of the atom relative to the size of the site. Small atoms are usually interstitial impurities, while larger atoms are usually substitutional impurities.
A vacancy is an atom site, normally occupied in the perfect crystal, from which an atom is missing. Often the term “vacancy” is used to denote a so-called Schottky defect, which is formed when an atom or an ion leaves a normal lattice site and repositions itself in a lattice site on the surface of the crystal. This may be the result of atomic rearrangement in an existing crystal at a high temperature when atomic mobility is high because of increased thermal vibrations. A vacancy may also originate in the process of crystallization as a result of local disturbances during the growth of new atomic planes on the crystal surface. Vacancies are point defects of a size nearly equal to the size of the original (occupied) site; the energy of the formation of a vacancy is relatively low - usually less than 1 eV.
The number of vacancies at equilibrium at each temperature in a crystal can be determined from eq. (2), in which \(\Delta \mathrm{H}_{\mathrm{d}}\) is the energy necessary to take an atom from a regular site of the crystal and place it on the surface for a Schottky-type defect. When a solid is heated a new higher equilibrium concentration of vacancies is established, usually first at crystal surfaces and then in the vicinity of dislocations and grain boundaries which provide sites for the atoms which have left their normal lattice site. Vacancies gradually spread throughout the crystal (from the surfaces into the bulk). On cooling the vacancy concentration is lowered by "diffusion of vacancies" to grain boundaries or dislocations, which act as sinks. In both cases, the new equilibrium vacancy concentration is established only after a finite amount of time. The rate at which vacancies move from point to point in the lattice decreases exponentially with decreasing temperature. Thus, on very rapid cooling (quenching) from a high temperature near the melting point most of the vacancies do not have time to diffuse to sinks and are said to be "frozen in". This gives a considerably greater ("non-equilibrium") concentration of vacancies in quenched specimens than that indicated by the thermal equilibrium value.
The concentration of vacant lattice sites in pure materials is very small at low temperatures - about one vacancy every \(10^8\) atom sites - and increases with increasing temperature to about one vacancy every \(10^3\) sites at the melting temperature. Vacancies are important because they control the rate of matrix (or substitutional) atom diffusion - i.e., atoms are able to move around in a crystalline solid primarily because of the presence of vacancies. (The mechanism by which they move is the same as that associated with moving a car in a filled parking lot to the exit). This is shown schematically in fig. 3. Self-interstitials are generally not encountered in close-packed

metallic systems, but may be introduced by irradiation. For example, high-energy neutrons from atomic fission can knock metal atoms from their regular sites into interstitial sites, creating vacancy-interstitial pairs.
Point Defects in Ionic Solids
Point defects in ionic structures differ from those found in pure elements because of the charge neutrality requirement. For example, in a pure monovalent ionic material a cation vacancy must have associated with it either a cation interstitial or an anion vacancy to maintain charge neutrality. Similar requirements hold for anion vacancies. A vacancy pair defect (migration of a cation and an anion to the surface) is usually called a Schottky imperfection, and a vacancy-interstitial pair defect is referred to as a Frenkel imperfection (an anion or cation has left its lattice position, which becomes a vacancy, and has moved to an interstitial position). These two types of imperfections are shown in fig. 4. Self-interstitials are much more common in ionic structures than in pure elements because many ionic compounds have relatively large interstitial sites available. That is, there are often interstitial sites in the unit cell that have nearly the

same surroundings as normal atom sites. (For example, in BeO the Be atoms fill only one-half the available tetrahedral sites, leaving four possible cation interstitial sites per unit cell. Thus a Be atom could go from a regular lattice site to an almost equivalent interstitial site with little distortion of the lattice.)
Foreign atoms in ionic crystals produce defects that also must maintain charge neutrality. For example, in NaCl a monovalent cation, such as lithium, may simply replace one of the sodium ions as a substitutional impurity. But a divalent cation, such as calcium, replacing a sodium ion must be accompanied by either a cation vacancy or an anion interstitial if charge neutrality is to be maintained. Correspondingly, monovalent impurity cations in a divalent structure (e.g., Na in MgO) must be accompanied by an appropriate number of cation interstitials or anion vacancies.
Point Defects in Covalently Bonded Solids
Substitutional impurities in covalently bonded materials can create a unique imperfection in the electronic structure if the impurity atom is from a group in the periodic table other than the matrix atoms. For example, you already considered Group V and Group III elements in a Group IV matrix, such as As or B in Si.
When foreign atoms are incorporated into a crystal structure, whether in substitutional or interstitial sites, we say that the resulting phase is a solid solution of the matrix material (solvent) and the foreign atoms (solute). The term “solid solution”, however, is not restricted to the low solute contents of doped semiconductor systems; there are many solid solutions, such as metallic alloys, that comprise a wide composition range.
LINE DEFECTS
Line imperfections, or dislocations, in crystalline solids are defects that produce lattice distortions centered about a line. A dislocation is simply the edge of an extra inserted fractional plane of atoms (fig. 5). Normally the symbol \(\perp\) is used to represent a positive
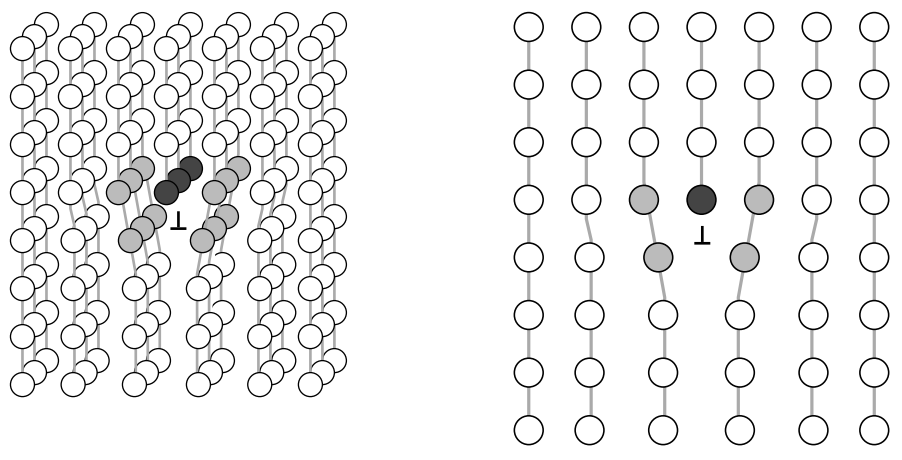
dislocation (extra fractional plane) and \(\top\) is used to represent a negative dislocation (missing fractional plane).
The importance of dislocations is readily demonstrated in the deformation of crystalline materials. The plane in which a dislocation moves through the lattice is called a slip plane. With an applied shear stress the dislocation moves, atomic row by atomic row, and one part of the crystal is displaced relative to the other. When the dislocation has passed through the crystal, the portion of the crystal above the slip plane has shifted one atomic distance relative to the portion below the slip plane. In other words, the motion of the dislocation has caused the crystal to change its shape - to be permanently deformed (fig. 6).

Please note: on either side of the dislocation the crystal lattice is essentially perfect, but in the immediate vicinity of the dislocation the lattice is severely distorted. For a positive edge dislocation, the presence of the extra half plane causes the atoms above the slip plane to be put in compression, while those below the slip plane are put in tension. Consequently, the edge dislocation will have a stress field around it that is compressive above the slip plane and tensile below the slip plane.
Plastic Deformation By Slip:
When single crystals of metal (or semiconductor) are pulled in tension, they will begin to deform (elongate) plastically at relatively low stress levels, and “blocks” of the crystals slide over one another because of dislocation motion. Simultaneously, so-called slip lines appear on their surface. It is found that deformation by slip occurs most easily on planes with high atomic density and with large interplanar spacing, while the direction of slip is in all instances an atomically “close-packed direction”. For FCC structures we therefore observe as the primary slip system {111} planes in direction, while in BCC structures the primary slip occurs on {110} planes in directions. (It should be noted that an alternate deformation mechanism is “deformation twinning”, presently not to be considered.)
Dislocation Climb:
Climb is the name given to the motion of dislocations when the extra “half” plane is extended farther into a crystal or partially withdrawn from it. Clearly, the climb process is not a motion of the plane, but rather its growth or shrinking as a result of the addition of atoms or “vacancies” respectively from the environment of the dislocation (fig. 7).

Multiplication of Dislocations:
Since during slip each dislocation leaves the matrix, macroscopic deformation could not take place given normal dislocation densities in the range of \(10^6-10^8 / \mathrm{cm}^3\). Examination of the deformed crystals indicates that multiplication of dislocations takes place during deformation. While there are a multitude of multiplication mechanisms, the one most extensively studied is the Frank-Read Source (not to be discussed in detail).
Dislocation Interactions:
The relative ease with which dislocations move across a solid matrix can be attributed to the severe displacements of atoms in the core of dislocations. If these local stresses are reduced, the mobility of dislocations - and thus the ease of slip - is reduced. It is found that impurities in the vicinity of dislocation cores tend to reduce the local distortion energy of the dislocations and thus stabilize the system against slip. In many systems impurities are intentionally added (e.g., solid solution hardening) to increase the strength of materials. Similarly, micro-precipitates tend to impede dislocation motion (e.g., precipitation hardening).
INTERFACIAL IMPERFECTIONS
The several different types of interfacial, or planar imperfections, in solids can be grouped into the following categories:
- Interfaces between solids and gases, which are called free surfaces;
- Interfaces between regions where there is a change in the electronic structure, but no change in the periodicity of atom arrangement, known as domain boundaries;
- Interfaces between two crystals or grains of the same phase where there is an orientation difference in the atom arrangement across the interface; these interfaces are called grain boundaries;
- Interfaces between different phases, called phase boundaries, where there is generally a change of chemical composition and atom arrangement across the interface.
Grain boundaries are peculiar to crystalline solids, while free surfaces, domain boundaries and phase boundaries are found in both crystalline and amorphous solids.
Free Surfaces
Because of their finite size, all solid materials have free surfaces. The arrangement of atoms at a free surface differs slightly from the interior structure because the surface atoms do not have neighboring atoms on one side. Usually the atoms near the surface have the same crystal structure but a slightly larger lattice parameter than the interior atoms.
Perhaps the most important aspect of free surfaces is the surface energy \((\gamma)\) associated with surfaces of any solid. The source of this surface energy may be seen by considering the surroundings of atoms on the surface and in the interior of a solid. To bring an atom from the interior to the surface, we must either break or distort some bonds - thereby increasing the energy. The surface energy is defined as the increase in energy per unit area of new surface formed. In crystalline solids, the surface energy depends on the crystallographic orientation of the surface - those surfaces that are planes of densest atomic packing are also the planes of lowest surface energy. This is because atoms on these surfaces have fewer of their bonds broken or, equivalently, have a larger number of nearest neighbors within the plane of the surface. Typical values of surface energies of solids range from about \(10^{-1}\) to \(1 \mathrm{~J} / \mathrm{m}^2\). Generally, the stronger the bonding in the crystal, the higher the surface energy.
Surface energies can be reduced by the adsorption of foreign atoms or molecules from the surrounding atmosphere. For example, in mica the surface energy of freshly cleaved material in a vacuum is much higher than the surface energy of the same surface cleaved in air. In this instance, oxygen is adsorbed from the air to partially satisfy the broken bonds at the surface. Impurity atom adsorption makes it almost impossible to maintain atomically clean surfaces. As a result, surface properties such as electron emission, rates of evaporation and rates of chemical reactions are extremely dependent on the presence of any adsorbed impurities. These properties will be different if the measurements are made under conditions giving different surface adsorption.
Grain Boundaries
Grain boundaries separate regions of different crystallographic orientation. The simplest form of a grain boundary is an interface composed of a parallel array of edge dislocations. This particular type of boundary is called a tilt boundary because the misorientation is in the form of a simple tilt about an axis, parallel to the dislocations. Tilt boundaries are referred to as low-angle boundaries because the angle of misorientation is generally less than \(10^{\circ}\).
When a grain boundary has a misorientation greater than \(10^{\circ}\) or \(15^{\circ}\), it is no longer practical to think of the boundary as being made up of dislocations because the spacing of the dislocations would be so small that they would lose their individual identity. The grain boundary represents a region a few atomic diameters wide where there is a transition in atomic periodicity between adjacent crystals or grains.
Grain boundaries have an interfacial energy because of the disruption in atomic periodicity in the vicinity of the boundary and the broken bonds that exist across the interface. The interfacial energy of grain boundaries is generally less than that of a free surface because the atoms in a grain boundary are surrounded on all sides by other atoms and have only a few broken or distorted bonds.
Solids with grain boundaries are referred to as polycrystalline, since the structure is composed of many crystals - each with a different crystallographic orientation. In the case of iron the grain boundary structure can be revealed by preferential chemical attack (etching) at the grain boundaries, while the grain structure in polyethylene is revealed by the use of polarized light. The grain structure is usually specified by giving average grain diameter or by using a scheme developed by the American Society for Testing and Materials (ASTM). In the ASTM procedure the grain size is specified by a “grain size number” (n) where
\[N=2^{n-1}\]
with \(\mathrm{N}\) equal to the number of grains per square inch when the sample is viewed at 100X magnification. For example, at a magnification of \(X=100\), a material with grain size number 8 will show 128 grains per inch \({ }^2\) - this material in effect has (at \(X=1\) ) \(1.28 \times 10^6\) grains per square inch. If the grains are approximately square in cross section, this corresponds to an average grain dimension of \(8.8 \times 10^{-4} \mathrm{in}^*\).
In polycrystalline samples the individual grains usually have a random crystallographic orientation with respect to one another, and the grain structure is referred to as randomly oriented. In some instances, however, the grains all have the same orientation to within a few degrees. In this instance the material is said to have a preferred orientation or texture.
Phase Boundaries
A phase is defined as a homogeneous, physically distinct and mechanically separable portion of the material with a given chemical composition and structure. Phases may be substitutional or interstitial solid solutions, ordered alloys or compounds, amorphous substances or even pure elements; a crystalline phase in the solid state may be either polycrystalline or exist as a single crystal.
Solids composed of more than one element may - and often do - consist of a number of phases. For example, a dentist's drill, something painfully familiar to all of us, consists of a mixture of small single crystals of tungsten carbide surrounded by a matrix of cobalt. Here the cobalt forms a continuous phase. Polyphase materials such as the dentist's drill are generally referred to as composite materials. Composite materials have great importance in the engineering world because they have many attractive properties that set them apart from single-phase materials. For example, the dentist’s drill has good abrasive characteristics (due to the hard carbide particles) and good toughness and impact resistance (due to the continuous cobalt matrix). Neither the tungsten carbide nor the cobalt has both abrasion resistance and impact resistance, yet the proper combination of the two phases yields a composite structure with the desired properties.
- *
-
ASTM has as yet not issued specifications in SI units!
The nature of the interface separating various phases is very much like a grain boundary. Boundaries between two phases of different chemical composition and different crystal structure are similar to grain boundaries, while boundaries between different phases with similar crystal structures and crystallographic orientations may be analogous to low-angle grain boundaries in both energy and structure.
The concept of a solid consisting of a continuous phase and a discontinuous phase (or phases) leads to a simple classification of the various types of composite materials. Table 1 gives this classification, which is based on the structure (whether amorphous or crystalline) of the continuous and discontinuous phases.
TABLE 1
Classification of Composite or Multiphase Materials
\(\begin{array}{lll}
\text { Continuous Phase } & \begin{array}{l}
\text { Discontinuous Phase } \\
\text { (or Phases) }
\end{array} & \text { Examples } \\
\hline \text { Crystalline } & \text { Crystalline } & \begin{array}{l}
\text { All metallic systems such as cast } \\
\text { iron, steel, soft solder, etc.; most } \\
\text { natural rocks such as granite and } \\
\text { marble. }
\end{array} \\ \\
\text { Crystalline } & \text { Amorphous } & \text { None of practical significance. } \\ \\
\text { Amorphous } & \text { Crystalline } & \begin{array}{l}
\text { Most man-made ceramics such } \\
\text { as building bricks and electrical } \\
\text { insulator porcelain, concrete, } \\
\text { partially crystalline polymers, } \\
\text { some polymer-crystalline particle } \\
\text { composites. }
\end{array} \\ \\
\text { Amorphous } & \text { Amorphous } & \begin{array}{l}
\text { Fiberglass, asphalt, wood, } \\
\text { hydrated cement, other gels. }
\end{array}
\end{array}\)