
18.5: Second Law and Gibbs Free Energy
- Page ID
- 60792

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Introduction
The Second Law of Thermodynamics states that a spontaneous process occurs when there is an increase in the entropy of the universe. Figure 18.5.1 shows a block diagram of the
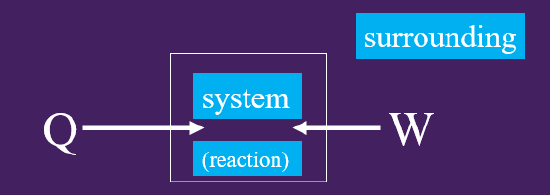
Figure \(\PageIndex{1}\): Block diagram of the universe consisting of the system (which is the reaction) and the surroundings.
Derivation of Gibbs Free Energy
According to the First Law of Thermodynamics, positive energy flow is when work is done on the system, or heat is added to it, and the total energy change of the universe is zero (the energy gained by the system is lost by the surroundings, and vice versa).
Writing the first law, and equating the entropy of the universe to that of the system plus the surroundings gives:
\[\Delta S_{Universe}>0 \\ \Delta S_{Universe}= \Delta S_{System} + \Delta S_{Surroundings} \\ \therefore \\ \Delta S_{System} + \Delta S_{Surroundings} >0\]
Noting that the system is the chemical reaction, and \(\Delta S_{reaction}\) can be determined from the standard molar entropies (see section 18.4.3.2, Entropies of Reaction), gives the following expression of the second law:
\[\large{\Delta S_{Reaction} + \Delta S_{Surroundings} >0}\]
Noting from the Clausius definition of entropy
\[\Delta S_{Surroundings}=\frac{q_{surroundings}}{T} \\ so \\ \large{\Delta S_{Reaction} + \frac{q_{Surroundings}}{T} >0} \]
and from the first law, that when the system loses heat, the surroundings gains heat,
\[q_{surroundings} = -q_{system} = -q_{reaction} \\ so \\ \large{\Delta S_{reaction} + \frac{-q_{reaction}}{T} >0} \]
The above equation is very important because we now define entropy change of the universe in terms of the system, ie., the chemical reaction, where it is the change in entropy of the reaction, plus the changed induced to the surrounding by the heat transferred, that is \(\frac{q}{T}\). Note, if the reaction is endothermic there is a reduction in entropy of the surroundings, and if it is exothermic, there is an increase.
Noting that the enthalpy of reaction is the heat transferred at constant pressure,
\[\Delta H_{reaction} = q_{p,reaction}\]
and substituting gives:
\[ \large{\textcolor{red}{\Delta S_{Reaction} + \frac{-\Delta H_{Reaction}}{T} >0}} \label{2nd}\]
Equation \ref{2nd} is very important because it allows us to describe the Second Law of Thermodynamics in terms of the system (chemical reaction), and not the universe. This allows us to use standard state thermodynamic tables of molar entropy and enthalpies of formation to determine the entropies and enthalpies of reaction, and use that to determine the direction a reaction will spontaneously proceed in.
This relationship is so important that it has been rearranged into a new equation, Gibbs Free Energy (G).
First, multiply everything by T, giving:
\[\large{T\Delta S_{reaction} -\Delta H_{reaction} >0 }\]
multiplying by negative 1 (-1) and rearranging (noting the sign of the inequality switches when you multiply by negative one).
\[\Delta H -T\Delta S <0\]
This is the definition of \(\Delta G\)
\[\large{\Delta G = \Delta H -T\Delta S}\]
And for a spontaneous reaction
\[\underbrace{\Delta G = \Delta H -T\Delta S <0}_{\large{\text{for a spontaneous reaction}}} \]
Gibbs Free Energy (G, sometimes given the symbol F), is also a state function, and all the things we learned with enthalpy in gen chem 1 apply. Its sign switches for the back reaction, it is an extensive property, and the values for a reaction can be determined from standard tables of free energies of formation.
Criteria for Spontaneity
Gibbs Free Energy quickly tells us if a system is at equilibrium \(\Delta G=0\), or if a reaction will occur (\(\Delta G \neq 0\)).

Figure\(\PageIndex{2}\): Spontaneity is expressed in terms of Gibbs Free Energy. Note, for a system at equilibria \(\Delta G = 0\), and if it is not at equilibria, either the forward or the reverse reaction will have a negative \(\Delta G\), and proceed spontaneously.
As pointed out in figure 18.5.3, Gibbs free energy has two terms that contribute to spontaneity, a temperature independent enthalpy term, and a temperature dependent entropy term.
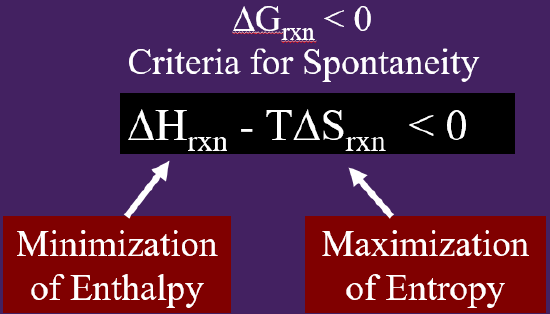
Figure\(\PageIndex{3}\): Gibbs Free Energy can be viewed as the results of two energetic processes that are a function of the system, Enthalpy minimization and the temperature dependent entropy maximization.
One could say from Gibbs Free Energy expression that from the perspective of the system, there are two driving "forces" towards spontaneity, a decrease in energy (exothermic reactions contribute to a negative \(\Delta G\)) and an increase in entropy (T is always positive, so only a [+] \(\Delta S\) would contribute to a negative \(\Delta G\) via the [-T\(\Delta S\)] term.)
| Type | \(\Delta H\) | \(\Delta S\) | \(\Delta G\) |
|---|---|---|---|
| 1. | [+] | [-] | Always [+], never spontaneous |
| 2. | [-] | [-] | [-] at low T, enthalpy driven process only spontaneous at low T |
| 3. | [+] | [+] | [-] at high T, entropy driven process only spontaneous at high T |
| 4. | [-] | [+] | Always [-], spontaneous at all T, both enthalpy and entropy driven |
Table\(\PageIndex{1}\), Temperature dependence of spontaneity as a function of the enthalpy and entropy changes of a reaction.
Phase Transitions
If a reaction is a phase change it fits the nature of types 3 & 4 in table 18.5.1. Melting is a type 3 process with a [+] \(\Delta H\) and \(\Delta S\) and is only spontaneous at a Temperature above the melting point. Freezing, the reverse process, is a type 2 process with a [-] \(\Delta H\) and [-] \(\Delta S\), and is only spontaneous below the freezing point. The freezing or melting point is when the solid and liquid phases can coexist, and at that temperature \(\Delta G =0\). The standard state freezing point can be calculated from thermodynamic tables. At the boiling point \( \Delta\)G=0 and the two phases can coexist in equilibrium.
\[\underbrace{\Delta G=\Delta H -T\Delta S = 0 }_{\text{at the freezing point}} \]
If we consider \(\Delta H\) and \(\Delta S\) to be independent of temperature we can substitute the standard values into the above equation and get:
\[\underbrace{\Delta G=\Delta H^o -T\Delta S^o = 0 }_{\text{at the freezing point}} \\ \therefore \\ T_{fp}=\frac{\Delta H^o_{s \rightarrow l}}{\Delta S^o_{s \rightarrow l}} \]
So the melting (or freezing) point is that temperature above which melting is spontaneous, below which freezing is spontaneous, and at which two phases can coexist in equilibrium. Thus you can use these tables to calculate phase transition temperatures like the melting or boiling point (next exercise).
Exercise \(\PageIndex{1}\)
Using Table T1, Standard Thermodynamic Quantities, calculate the boiling point of ethanol, C2H5OH .
- Answer
-
First calculate the entropy and enthalpy change associated with boiling from the standard state molar entropies and enthalpies of formation
\[\Delta H^o_{boiling} = \Delta H^o_f(gas) - \Delta H^o_f(liq) = -234.8-(-277.6)=42.8\frac{kJ}{mol} \nonumber \\ \Delta S^o_{boiling} = \Delta S^o_f(gas) - \Delta S^o_f(liq) = 281.6-160.7=120.9\frac{J}{mol \cdot K} \nonumber \]
Then noting that at the boiling point the two phases can coexist in equilibrium.
\[\Delta G^o=\Delta H^o -T\Delta S^o = 0 \nonumber \]
Rearranging and solving for T.
\[T=\frac{\Delta H^o_{boiling}}{\Delta S^o_{boiling}}=\frac{42.8 \frac{kJ}{mol}} {0.1209\frac{kJ}{mol \cdot K}}=354K = 80.9^oC \nonumber \]
Driving Chemical Reactions
Gibbs Free Energy describes the spontaneity of a chemical reaction in terms of the entropy and enthalpy changes within the system, that is, of the chemical reaction itself. In section 15.2.5 the reaction quotient was introduced, which was the equilibrium constant expression at any conditions, and if \(Q \neq K), the system was not at equilibrium, and a reaction would spontaneously proceed in either the forward or reverse direction, depending on if Q is less than, or greater than K.
Q=K, No reaction, system is at equilibrium
Q>K, Product loaded, reactants are produced until Q=K
Q<K, Reactant loaded, products are produced until Q=K
This can be equated to the free energy through the following equation
\[\large{\Delta G = \Delta G^o +RTlnQ}\]
This is an important equation because it allows thermodynamics to relate spontaneity to reaction concentrations. That is, there are three possible conditions, the system is at equilibrium and no reaction occurs, it is Reactant Loaded and the Forward Reaction occurs, or it is Product Loaded, and the back reaction occurs.
Equilibrium Conditions
At equilibrium Q=K and \(\Delta G_{rxn} \) = 0, so substituting these equilibrium conditions into eq. 18.5.11 gives
\[0=\Delta G^o_{rxn} + RTlnK_{eq}\]
Rearranging gives two important equations:
\[\Delta G^o_{rxn} = -RTlnK\]
and
\[\large{\textcolor{red}{K=e^{\frac{-\Delta G^o_{rxn}}{RT}}}}\]
Equation 18.5.14 is very important because it allows us to calculate the equilibrium constant from standard state values. Note, if you are calculating K at 25o C you can calculate \(\Delta G^o_{rxn}\) from \(\Delta G^o_f\) in the standard thermodynamic tables.
\[ \Delta G^o_{reaction} = \sum n\Delta G^o_{f_{products}} - \sum m\Delta G^o_{f_{reactants}} \\ \text{ (Where m and n are the respective stoichiometric coefficients of the reactant and product species.)} \]
Reactant Loaded Systems
In a Reactant loaded system Q<K (review section 15.2.5) and the forward reaction occurs until Q=K. We will now show that when Q<K the value of \(\Delta G \; becomes <0\).
Starting with
\[\large{\Delta G = \Delta G^o +RTlnQ}\]
and substituting
\[\large{\Delta G^o =-RTlnK}\]
gives
\[\large{\Delta G = -RTlnK +RTlnQ}\]
Which can be rearranged to
\[\large{\Delta G = RTln \frac{Q}{K}}\]
If Q<K, then \(\frac{Q}{K}\) is a fraction and its natural log is a negative number. Since R is a positive constant, and T is an absolute temperature (there are no negative degrees Kelvin), then \(RTln\frac{Q}{K}\) must be negative, and thus \(\Delta G\) is negative.
Product Loaded Systems
Here Q>K and the back reaction occurs until Q=K. We can use the exact same logic as above, and in eq. 18.5.20, except that \(\frac{Q}{K}\) is a number bigger than one, which means its log is a positive value. This may be easier to see by rewriting eq. 18.5.20 into its exponential form
\[\large{\frac{Q}{K}=e^{\frac{\Delta G}{RT}}}\]
noting if \(\Delta G = 0 \; then \; e^{\frac{\Delta G}{RT}}= e^o =1\) and Q=K
if \(\Delta G > 0 \; then \; e^{\frac{\Delta G}{RT}}= e^{[\text{positive number}]} >1\) and Q>K , and the back reaction occurs until Q=K and \(\Delta G = 0\)
Likewise, for a Reactant loaded system (above)
\(\Delta G < 0 \; then \; e^{\frac{\Delta G}{RT}}= e^{[\text{negative number}]} <1\) (a fraction) and Q<K, and the forward reaction occurs until Q=K and \(\Delta G = 0 \)
Contributors and Attributions
Robert E. Belford (University of Arkansas Little Rock; Department of Chemistry). The breadth, depth and veracity of this work is the responsibility of Robert E. Belford, rebelford@ualr.edu. You should contact him if you have any concerns. This material has both original contributions, and content built upon prior contributions of the LibreTexts Community and other resources, including but not limited to: