
26.10: Protein Structure
- Page ID
- 36475
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\(\newcommand{\ket}[1]{\left| #1 \right>} \)
\( \newcommand{\bra}[1]{\left< #1 \right|} \)
\( \newcommand{\braket}[2]{\left< #1 \vphantom{#2} \right| \left. #2 \vphantom{#1} \right>} \)
\( \newcommand{\qmvec}[1]{\mathbf{\vec{#1}}} \)
\( \newcommand{\op}[1]{\hat{\mathbf{#1}}}\)
\( \newcommand{\expect}[1]{\langle #1 \rangle}\)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)After completing this section, you should be able to
- discuss, with reference to a suitable example (either given or of your own choice), the structure of proteins, paying particular attention to distinguishing between the primary, secondary, tertiary and quaternary structure.
- describe the α‑helical secondary structure displayed by many proteins.
- describe the β‑pleated‑sheet structure displayed by many proteins.
Make certain that you can define, and use in context, the key terms below.
- α helix
- β pleated sheet
- primary structure
- quaternary structure
- secondary structure
- tertiary structure
Note that in a diagram of the α‑helical structure of a protein, the C‑terminal of the protein is at the bottom of the diagram and the N‑terminal is at the top. In an α helix, such as the one shown in Figure 26.9.1, the bulky R groups are all found on the outside of the helix, where they have the most room.
The four levels of protein structure
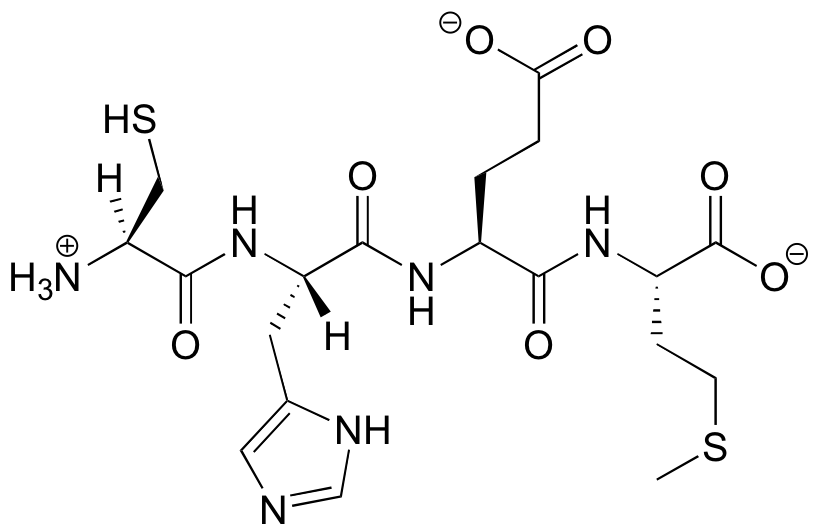
Protein structure can be discussed at four distinct levels. A protein’s primary structure is two-dimensional - simply the sequence of amino acids in the peptide chain. Below is a Lewis structure of a short segment of a protein with the sequence CHEM (cysteine - histidine - glutamate - methionine)
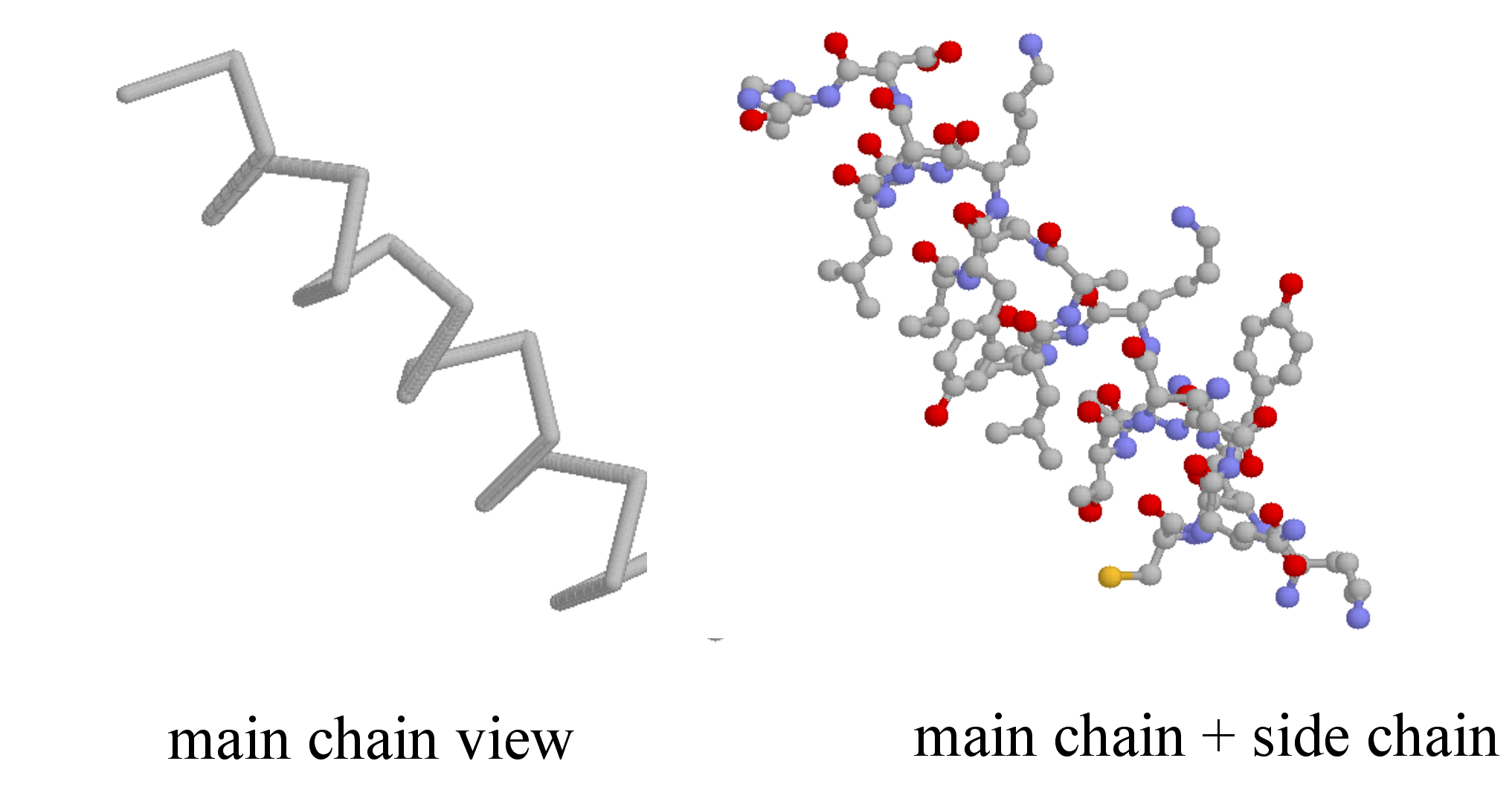
Secondary structure is three-dimensional, but is a local phenomenon, confined to a relatively short stretch of amino acids. For the most part, there are three important elements of secondary structure: helices, beta-sheets, and loops. In a helix, the main chain of the protein adopts the shape of a clockwise spiral staircase, and the side chains point out laterally.
In a beta-sheet (or beta-strand) structure, two sections of protein chain are aligned side-by-side in an extended conformation. The figure below shows two different views of the same beta-sheet: in the left-side view, the two regions of protein chain are differentiated by color.

Loops are relatively disordered segments of protein chain, but often assume a very ordered structure when in contact with a second protein or a smaller organic compound.
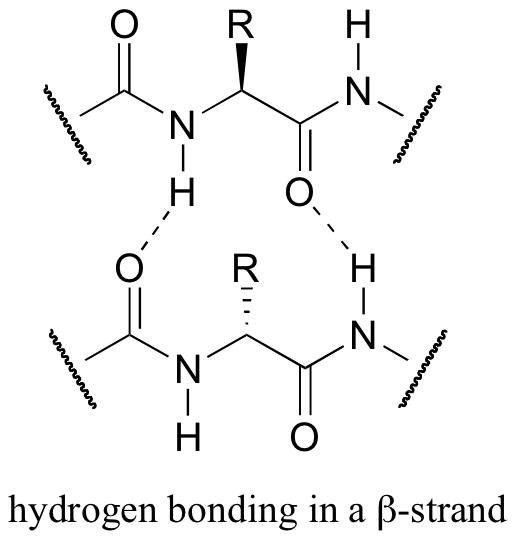
Both helix and the beta-sheet structures are held together by very specific hydrogen-bonding interactions between the amide nitrogen on one amino acid and the carbonyl oxygen on another. The hydrogen bonding pattern in a section of a beta-strand is shown below.
Secondary structure refers to the shape of a folding protein due exclusively to hydrogen bonding between its backbone amide and carbonyl groups. Secondary structure does not include bonding between the R-groups of amino acids, hydrophobic interactions, or other interactions associated with tertiary structure. The two most commonly encountered secondary structures of a polypeptide chain are α-helices and beta-pleated sheets. These structures are the first major steps in the folding of a polypeptide chain, and they establish important topological motifs that dictate subsequent tertiary structure and the ultimate function of the protein.
α-Helices
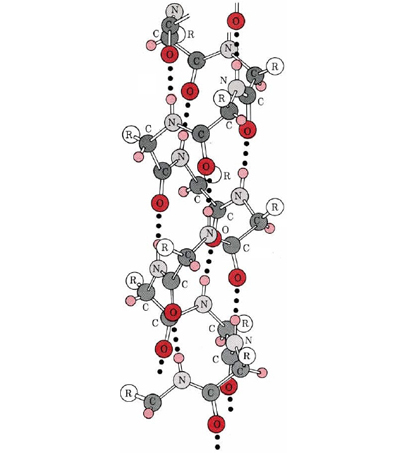
An α-helix is a right-handed coil of amino-acid residues on a polypeptide chain, typically ranging between 4 and 40 residues. This coil is held together by hydrogen bonds between the oxygen of C=O on top coil and the hydrogen of N-H on the bottom coil. Such a hydrogen bond is formed exactly every 4 amino acid residues, and every complete turn of the helix is only 3.6 amino acid residues. This regular pattern gives the α-helix very definite features with regards to the thickness of the coil and the length of each complete turn along the helix axis.
The structural integrity of an α-helix is in part dependent on correct steric configuration. Amino acids whose R-groups are too large (tryptophan, tyrosine) or too small (glycine) destabilize α-helices. Proline also destabilizes α-helices because of its irregular geometry; its R-group bonds back to the nitrogen of the amide group, which causes steric hindrance. In addition, the lack of a hydrogen on Proline's nitrogen prevents it from participating in hydrogen bonding.
Another factor affecting α-helix stability is the total dipole moment of the entire helix due to individual dipoles of the C=O groups involved in hydrogen bonding. Stable α-helices typically end with a charged amino acid to neutralize the dipole moment.
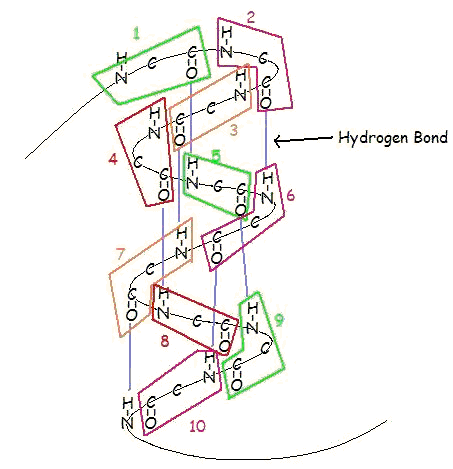
BETA-PLEATED SHEETS
This structure occurs when two (or more, e.g. ψ-loop) segments of a polypeptide chain overlap one another and form a row of hydrogen bonds with each other. This can happen in a parallel arrangement:

Or in anti-parallel arrangement:
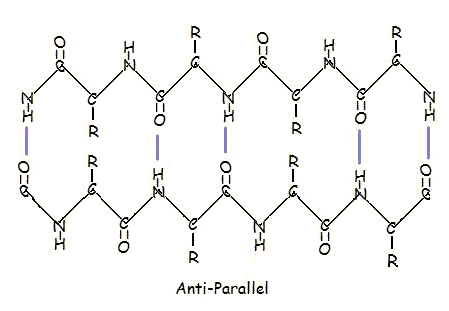
Parallel and anti-parallel arrangement is the direct consequence of the directionality of the polypeptide chain. In anti-parallel arrangement, the C-terminus end of one segment is on the same side as the N-terminus end of the other segment. In parallel arrangement, the C-terminus end and the N-terminus end are on the same sides for both segments. The "pleat" occurs because of the alternating planes of the peptide bonds between amino acids; the aligned amino and carbonyl group of each opposite segment alternate their orientation from facing towards each other to facing opposite directions.
The parallel arrangement is less stable because the geometry of the individual amino acid molecules forces the hydrogen bonds to occur at an angle, making them longer and thus weaker. Contrarily, in the anti-parallel arrangement the hydrogen bonds are aligned directly opposite each other, making for stronger and more stable bonds.
Commonly, an anti-parallel beta-pleated sheet forms when a polypeptide chain sharply reverses direction. This can occur in the presence of two consecutive proline residues, which create an angled kink in the polypeptide chain and bend it back upon itself. This is not necessary for distant segments of a polypeptide chain to form beta-pleated sheets, but for proximal segments it is a definite requirement. For short distances, the two segments of a beta-pleated sheet are separated by 4+2n amino acid residues, with 4 being the minimum number of residues.
α-PLEATED SHEETS
A similar structure to the beta-pleated sheet is the α-pleated sheet. This structure is energetically less favorable than the beta-pleated sheet, and is fairly uncommon in proteins. An α-pleated sheet is characterized by the alignment of its carbonyl and amino groups; the carbonyl groups are all aligned in one direction, while all the N-H groups are aligned in the opposite direction. The polarization of the amino and carbonyl groups results in a net dipole moment on the α-pleated sheet. The carbonyl side acquires a net negative charge, and the amino side acquires a net positive charge.

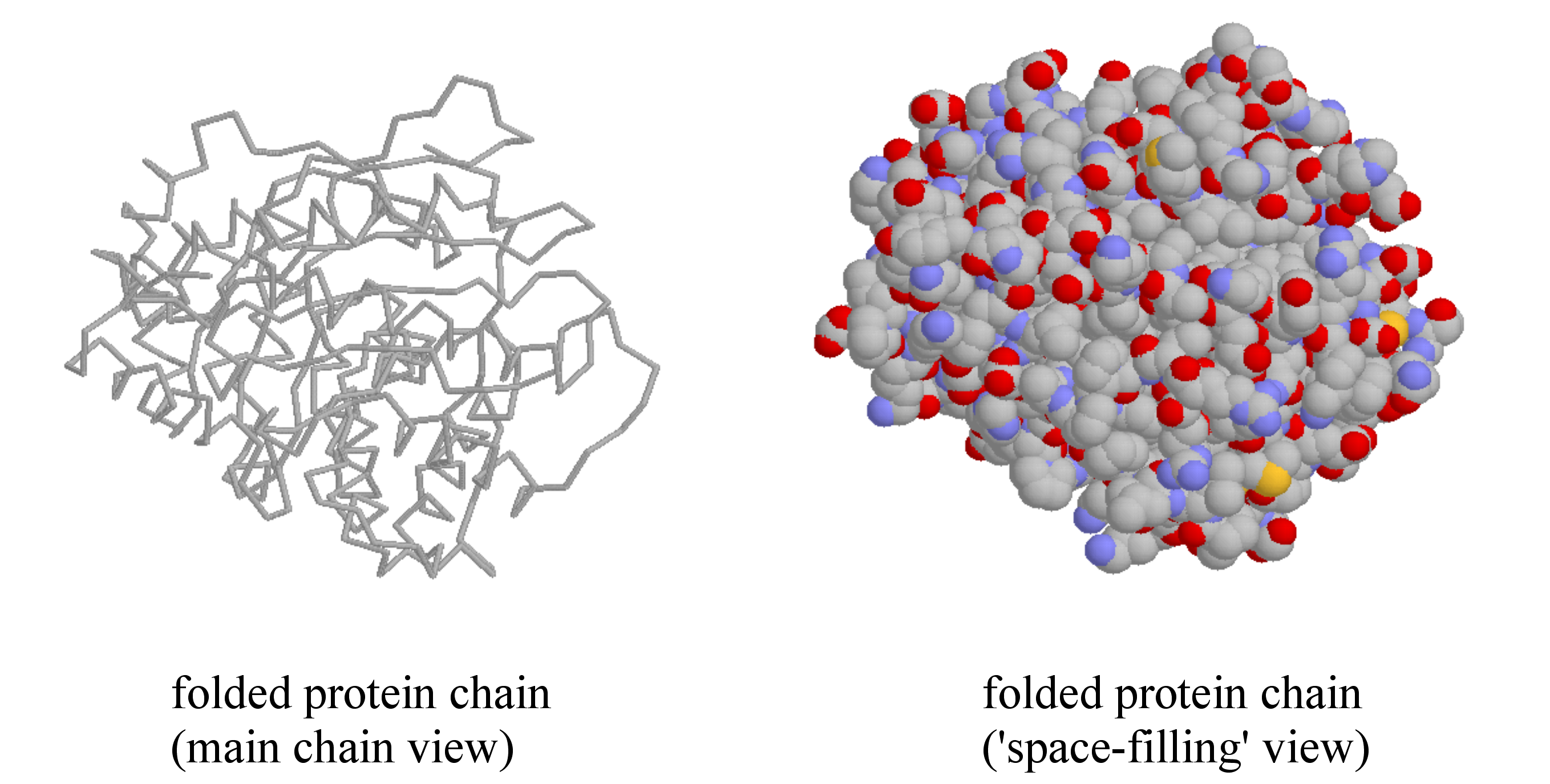
A protein’s tertiary structure is the shape in which the entire protein chain folds together in three-dimensional space, and it is this level of structure that provides protein scientists with the most information about a protein’s specific function.
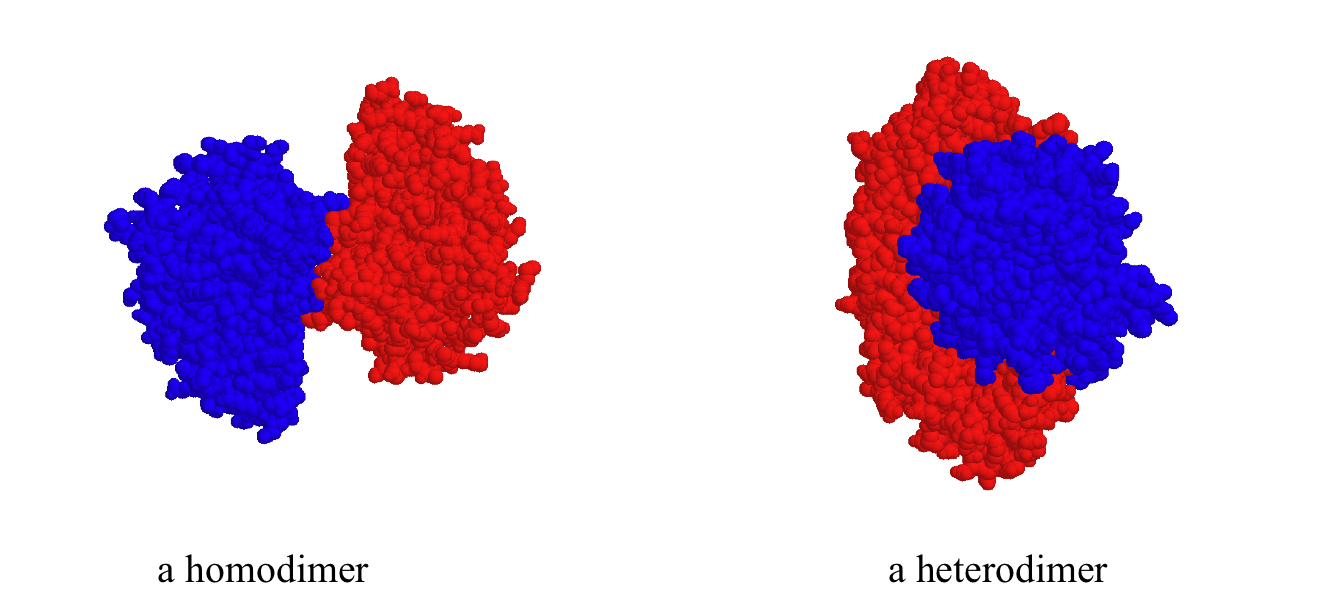
While a protein's secondary and tertiary structure is defined by how the protein chain folds together, quaternary structure is defined by how two or more folded protein chains come together to form a 'superstructure'. Many proteins consist of only one protein chain, or subunit, and thus have no quaternary structure. Many other proteins consist of two identical subunits (these are called homodimers) or two non-identical subunits (these are called heterodimers).
Quaternary structures can be quite elaborate: below we see a protein whose quaternary structure is defined by ten identical subunits arranged in two five-membered rings, forming what can be visualized as a 'double donut' shape (this is fructose 1,6-bisphosphate aldolase):

The molecular forces that hold proteins together
The question of exactly how a protein ‘finds’ its specific folded structure out of the vast number of possible folding patterns is still an active area of research. What is known, however, is that the forces that cause a protein to fold properly and to remain folded are the same basic noncovalent forces that we talked about in chapter 2: ion-ion, ion-dipole, dipole-dipole, hydrogen bonding, and hydrophobic (van der Waals) interactions. One interesting type of hydrophobic interaction is called ‘aromatic stacking’, and occurs when two or more planar aromatic rings on the side chains of phenylalanine, tryptophan, or tyrosine stack together like plates, thus maximizing surface area contact.
Hydrogen bonding networks are extensive within proteins, with both side chain and main chain atoms participating. Ionic interactions often play a role in protein structure, especially on the protein surface, as negatively charged residues such as aspartate interact with positively-charged groups on lysine or arginine.
One of the most important ideas to understand regarding tertiary structure is that a protein, when properly folded, is polar on the surface and nonpolar in the interior. It is the protein's surface that is in contact with water, and therefore the surface must be hydrophilic in order for the whole structure to be soluble. If you examine a three dimensional protein structure you will see many charged side chains (e.g. lysine, arginine, aspartate, glutamate) and hydrogen-bonding side chains (e.g. serine, threonine, glutamine, asparagine) exposed on the surface, in direct contact with water. Inside the protein, out of contact with the surrounding water, there tend to be many more hydrophobic residues such as alanine, valine, phenylalanine, etc. If a protein chain is caused to come unfolded (through exposure to heat, for example, or extremes of pH), it will usually lose its solubility and form solid precipitates, as the hydrophobic residues from the interior come into contact with water. You can see this phenomenon for yourself if you pour a little bit of vinegar (acetic acid) into milk. The solid clumps that form in the milk are proteins that have come unfolded due to the sudden acidification, and precipitated out of solution.
In recent years, scientists have become increasing interested in the proteins of so-called ‘thermophilic’ (heat-loving) microorganisms that thrive in hot water environments such as geothermal hot springs. While the proteins in most organisms (including humans) will rapidly unfold and precipitate out of solution when put in hot water, the proteins of thermophilic microbes remain completely stable, sometimes even in water that is just below the boiling point. In fact, these proteins typically only gain full biological activity when in appropriately hot water - at room-temperature they act is if they are ‘frozen’. Is the chemical structure of these thermostable proteins somehow unique and exotic? As it turns out, the answer to this question is ‘no’: the overall three-dimensional structures of thermostable proteins look very much like those of ‘normal’ proteins. The critical difference seems to be simply that thermostable proteins have more extensive networks of noncovalent interactions, particularly ion-ion interactions on their surface, that provides them with a greater stability to heat. Interestingly, the proteins of ‘psychrophilic’ (cold-loving) microbes isolated from pockets of water in arctic ice show the opposite characteristic: they have far fewer ion-ion interactions, which gives them greater flexibility in cold temperatures but leads to their rapid unfolding in room temperature water.