
17.6: Reactions of Alcohols
- Page ID
- 36350
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)After completing this section, you should be able to
- discuss the reactions of alcohols that have been introduced in previous units. These reactions include
- conversion of alcohols into alkyl halides.
- conversion of alcohols into tosylates.
- dehydration of alcohols to yield alkenes.
- conversion of alcohols into esters.
As you read through Section 17.6 you should be prepared to turn back to those earlier sections in which some of the reactions of alcohols were discussed:
- dehydration to alkenes—Section 8.1.
- conversion to alkyl halides—Section 10.5.
You may also wish to review the discussion of acidity constants, which can be found in Section 2.8.
Remember that when an alcohol reacts with tosyl chloride to form a tosylate, it is the O-H bond of the alcohol that is broken, not the C-O bond. This means that the absolute configuration of the carbon atom attached to the hydroxyl group remains unchanged throughout the reaction. The reading illustrates how this fact can be exploited to control the stereochemistry in an organic synthesis.
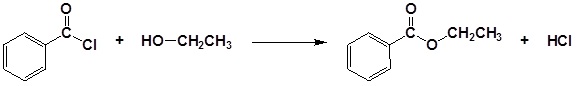
Finally, the reading shows the production of an ester from an alcohol and and an acid chloride. In Section 21.3 we will discuss the Fischer esterification, a famous reaction that uses an alcohol and a carboxylic acid to form the ester.
Conversion of Alcohols into Alkyl Halides
When alcohols react with a hydrogen halide, a substitution takes place producing an alkyl halide and water:
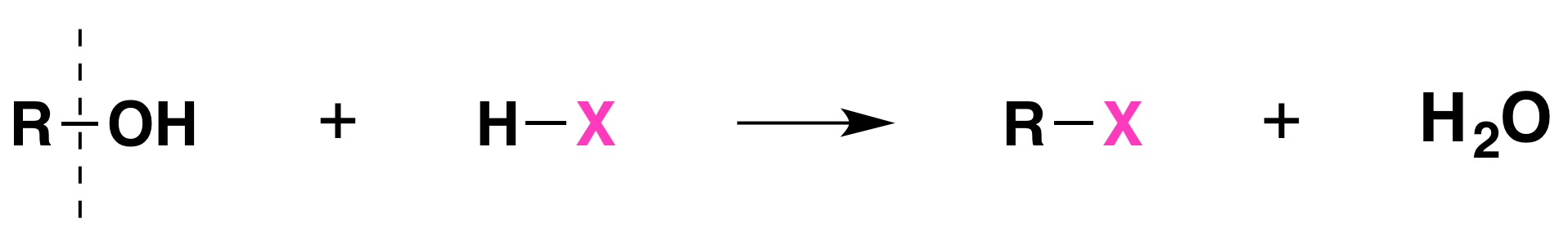
- The order of reactivity of alcohols is 3° > 2° > 1° methyl.
- The order of reactivity of the hydrogen halides is HI > HBr > HCl (HF is generally unreactive).
The reaction is acid catalyzed. Alcohols react with the strongly acidic hydrogen halides HCl, HBr, and HI, but they do not react with nonacidic NaCl, NaBr, or NaI. Primary and secondary alcohols can be converted to alkyl chlorides and bromides by allowing them to react with a mixture of a sodium halide and sulfuric acid:
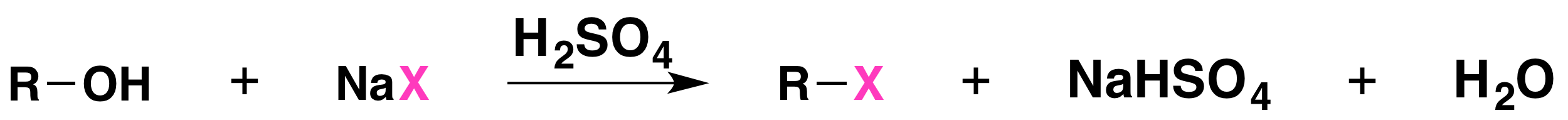
Mechanisms of the Reactions of Alcohols with HX
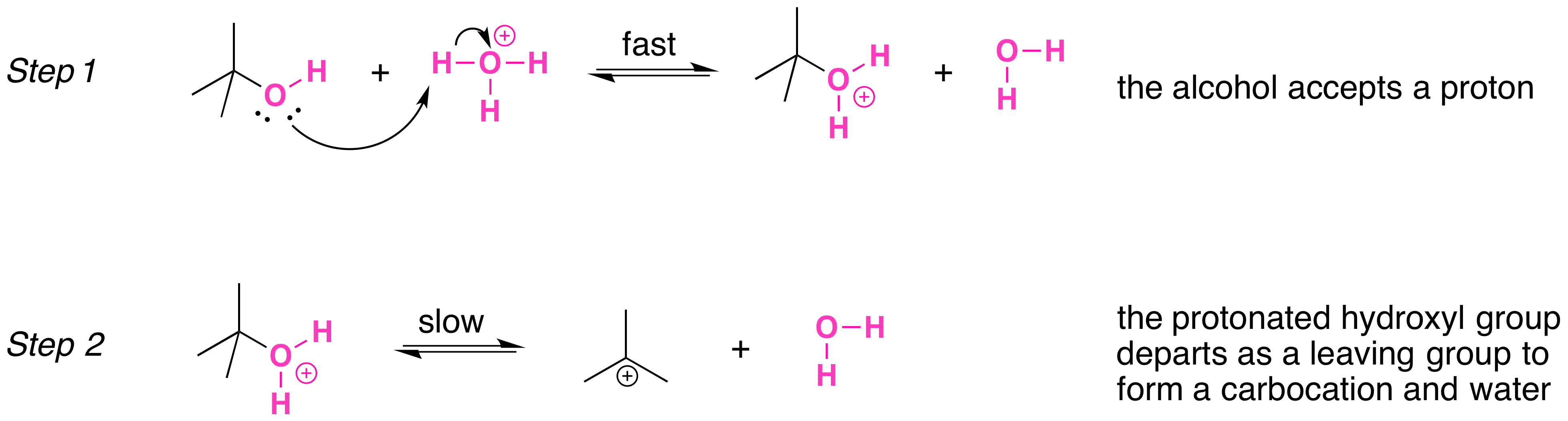
Secondary, tertiary, allylic, and benzylic alcohols appear to react by a mechanism that involves the formation of a carbocation, in an SN1 reaction with the protonated alcohol acting as a leaving group.
The SN1 mechanism is illustrated by the reaction of tert-butyl alcohol and aqueous hydrochloric acid (H3O+, Cl- ). The first two steps in this SN1 substitution mechanism are protonation of the alcohol to form an oxonium ion. Although the oxonium ion is formed by protonation of the alcohol, it can also be viewed as a Lewis acid-base complex between the cation R+ and H2O. Protonation of the alcohol converts a poor leaving group (OH-) to a good leaving group H2O, which makes the dissociation step of the SN1 mechanism more favorable.
In step 3, the carbocation reacts with a nucleophile (a halide ion) to complete the substitution.

When we convert an alcohol to an alkyl halide, we carry out the reaction in the presence of acid and in the presence of halide ions, and not at elevated temperature. Halide ions are good nucleophiles (they are much stronger nucleophiles than water), and since halide ions are present in high concentration, most of the carbocations react with an electron pair of a halide ion to form a more stable species, the alkyl halide product. The overall result is an SN1 reaction.
Not all acid-catalyzed conversions of alcohols to alkyl halides proceed through the formation of carbocations. Primary alcohols and methanol react to form alkyl halides under acidic conditions by an SN2 mechanism. In these reactions the function of the acid is to produce a protonated alcohol. The halide ion then displaces a molecule of water (a good leaving group) from carbon; this produces an alkyl halide:
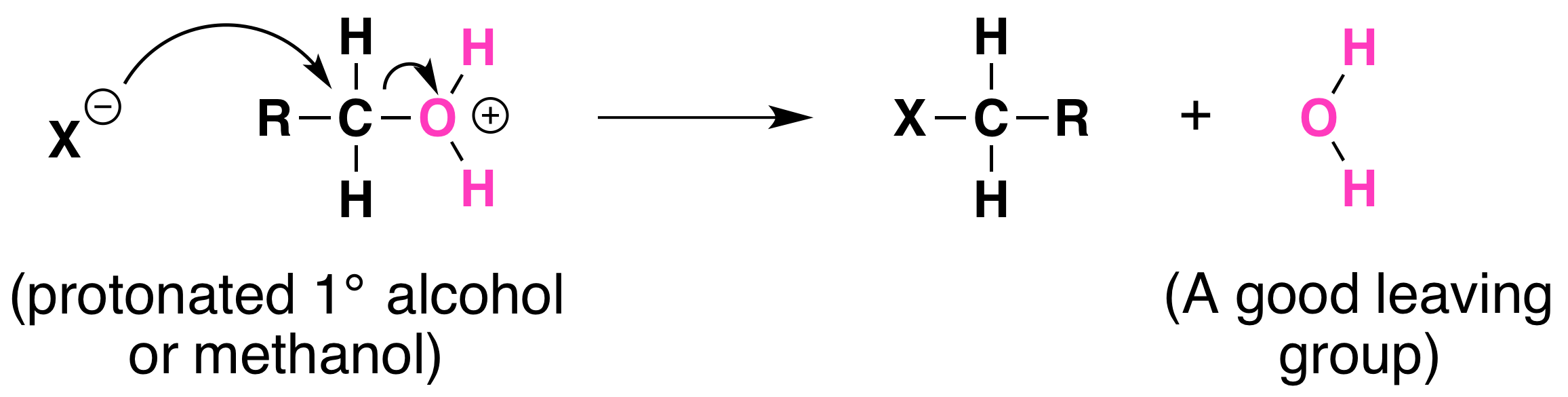
Again, acid is required. Although halide ions (particularly iodide and bromide ions) are strong nucleophiles, they are not strong enough to carry out substitution reactions with alcohols themselves. Direct displacement of the hydroxyl group does not occur because the leaving group would have to be a strongly basic hydroxide ion:

Conversion of Alcohols into Alkyl Halides Using SOCl2 or PBr3
The most common methods for converting 1º- and 2º-alcohols to the corresponding chloro and bromo alkanes (i.e. replacement of the hydroxyl group) are treatments with thionyl chloride and phosphorus tribromide, respectively. These reagents are generally preferred over the use of concentrated HX due to the harsh acidity of these hydrohalic acids and the carbocation rearrangements associated with their use.
Synthetic organic chemists, when they want to convert an alcohol into a better leaving group, have several methods to choose from. One common strategy is to convert the alcohol into an alkyl chloride or bromide, using thionyl chloride or phosphorus tribromide:
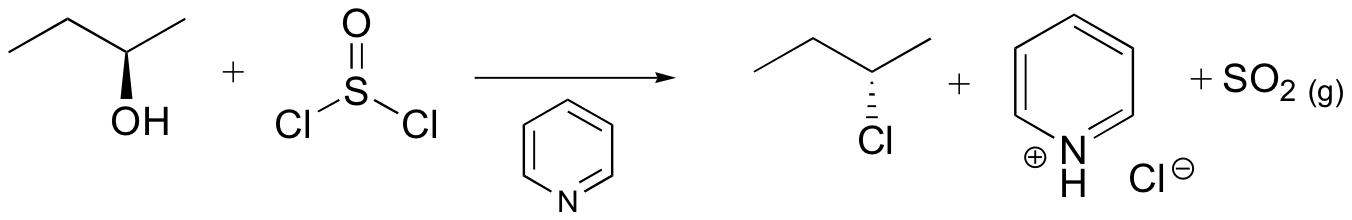
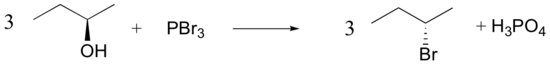
Mechanisms
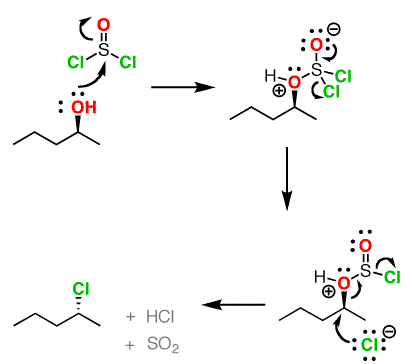
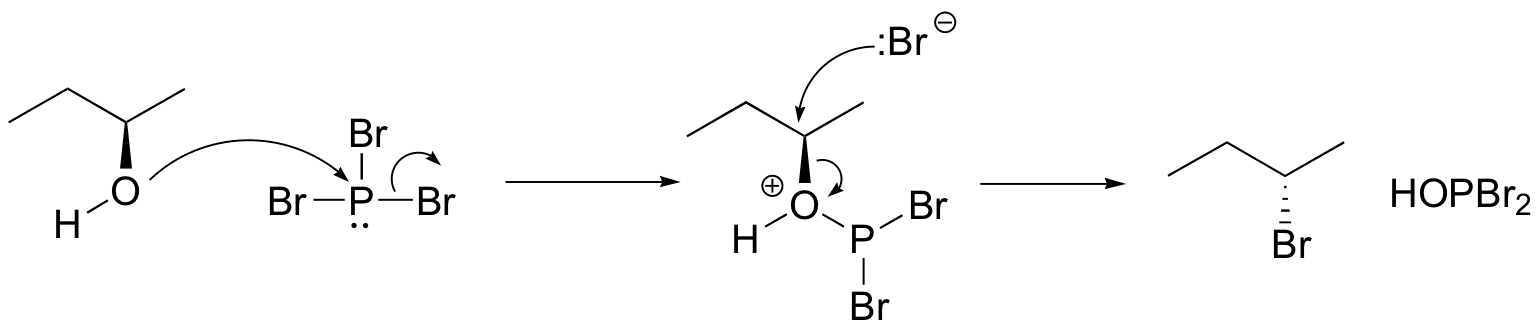
Both of these reagents form an alkyl halide through an SN2 mechanism. The mechanism for both reactions start by making the alcohol's -OH a better leaving group through conversion to an intermediate. Thionyl chloride creates an intermediate chlorosulfite (-OSOCl2) compound and phosphorus tribromide makes an intermediate dibromophosphite (-OPBr2) compound. These intermediate compounds can subsequently be eliminated as a leaving group during an SN2 reaction with the corresponding nucleophilic halide ion. Since these reactions proceeds through a backside attack, there is inversion of configuration at the carbon.
Thionyl Chloride
Notice that during the reaction with thonyl chloride hydrochloric acid (HCl) and sulfurdioxide (SO2) are produced as byproducts.
Phosphorustribromide
During this reaction HOPBr2 is made as biproducts.
Conversion of Alcohols into Tosylates
Alternatively, we can transform an alcohol group into sulfonic ester using para-toluene sulfonyl chloride (Ts-Cl) or methanesulfonyl chloride (Ms-Cl), creating what is termed an organic tosylate or mesylate. Notice that unlike the halogenation reactions above, the C-O bond of the alcohol is not broken during the conversion to a tosylate or mesylate so the reaction proceeds with retention of configuration at the electrophilic carbon.
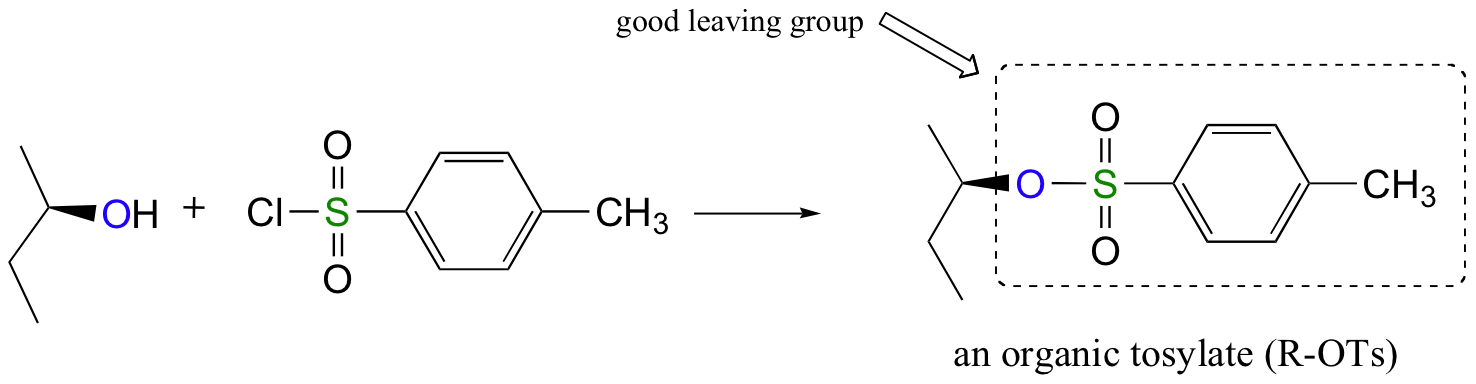
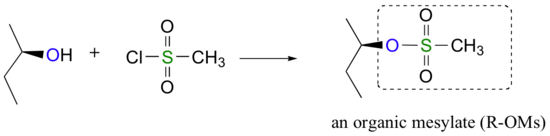
Tosylate and mesylate groups are excellent leaving groups in nucleophilic substitution reactions, due to resonance delocalization of the developing negative charge on the leaving oxygen. They behave much line alkyl halides and can undergo SN2 and SN1 reaction depending on the conditions.

The laboratory synthesis of isopentenyl diphosphate - the 'building block' molecule used by nature for the construction of isoprenoid molecules such as cholesterol and b-carotene - was accomplished by first converting the alcohol into an organic tosylate (step 1), then displacing the tosylate group with an inorganic pyrophosphate nucleophile (step 2) (J. Org. Chem 1986, 51, 4768).
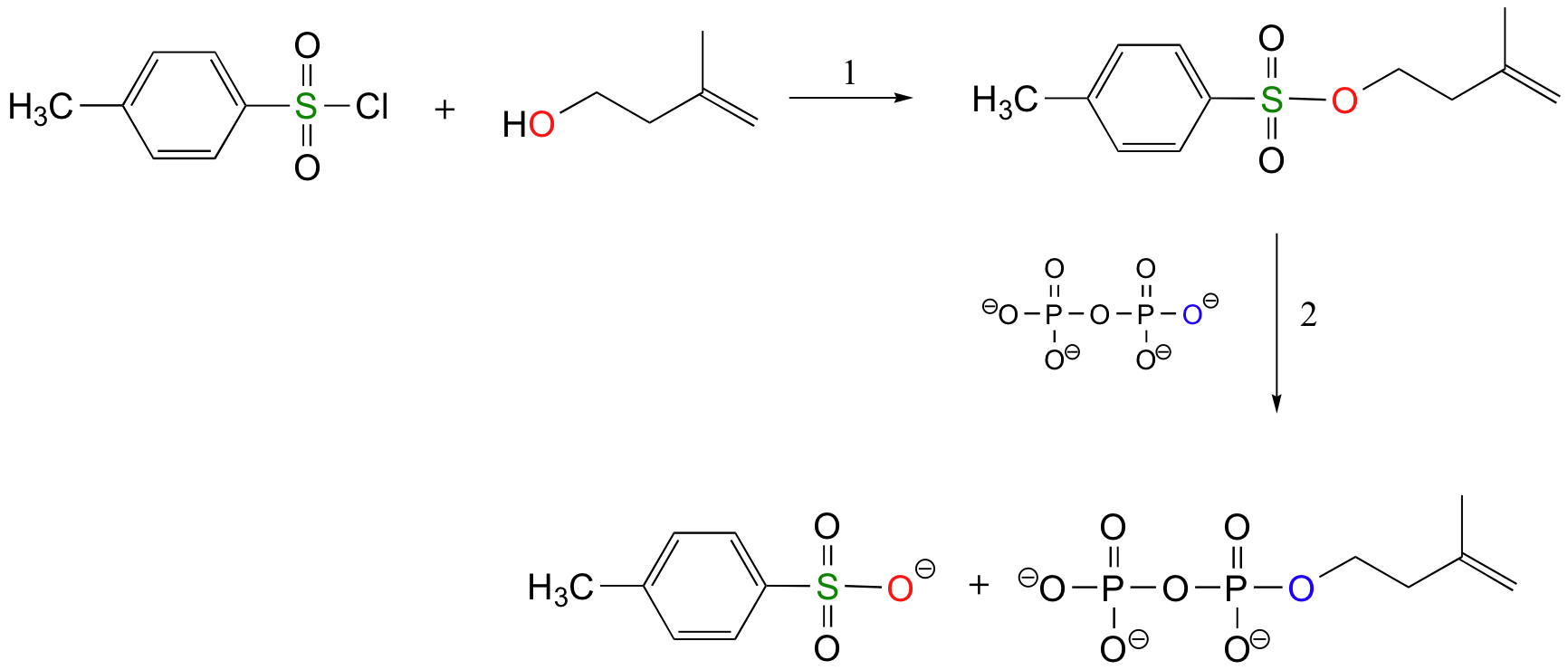
Using Sulfonate Esters for Stereochemical Control
Tosylate and mesylate group's retention of conversion during formation makes them an important source of stereochemical control in organic synthesis. In the conversion of an alcohol to a halide for a subsequent SN2 substitution there are two inversions of configuration. One during the conversion of the alcohol to a halide and the second during the SN2 substitution reaction. Overall, these reaction steps produce a product with the same stereochemistry as the alcohol starting material.
When a tosylate or a mesylate are used for a similar conversion there is only one inversion of configuration. During the conversion of an alcohol to a tosylate the configuration of the alcohol strarting material is retained. This means that the tosylate will have the same stereochemical configuration as the alcohol starting material. The subsequent SN2 reaction with the tosylate causes an inversion of configuration which provides a product of opposite stereochemistry as the alcohol starting material. The figure below outlines the relative stereochemistry of a series of reactions which summarize these points.

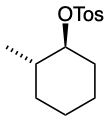
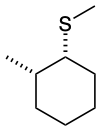
Predict the structures of A and B in the following reaction:
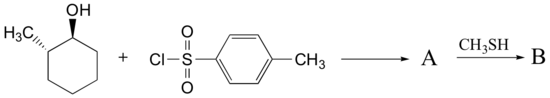
A. The tosylate is formed with retained stereochemistry.
B. The thioether is formed with inverted stereochemistry.
Dehydration of Alcohols to Yield Alkenes
One way to synthesize alkenes is by dehydration of alcohols, a process in which alcohols undergo E1 or E2 mechanisms to lose water and form a double bond. The dehydration reaction of alcohols to generate alkene proceeds by heating the alcohols in the presence of a strong acid, such as sulfuric or phosphoric acid, at high temperatures.
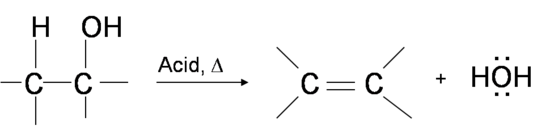
The required range of reaction temperature decreases with increasing substitution of the hydroxy-containing carbon:
- 1° alcohols: 170° - 180°C
- 2° alcohols: 100°– 140 °C
- 3° alcohols: 25°– 80°C
If the reaction is not sufficiently heated, the alcohols do not dehydrate to form alkenes, but react with one another to form ethers (e.g., the Williamson Ether Synthesis).
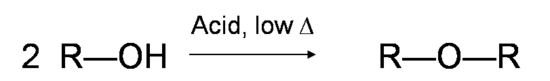
Mechanism for the Dehydration of Alcohol into Alkene
Primary alcohols undergo bimolecular elimination (E2 mechanism) while secondary and tertiary alcohols undergo unimolecular elimination (E1 mechanism). The relative reactivity of alcohols in dehydration reaction is ranked as the following
Methanol < primary < secondary < tertiary
Primary alcohol dehydrates through the E2 mechanism. First, the hydoxyl oxygen becomes protonated by reagent acid (sulfuric acid H2SO4), forming an alkyloxonium ion leaving group. Next, in a concerted process, then the conjugate base of the reagent acid ( HSO4–) removes an adjacent hydrogen, the alkyloxonium ion is removed as a leaving group, and alkene double bond is formed. Remember that this reaction follows Zaitsev's rule which states that the most stable alkene will be the major product. In this reaction there is only one possible alkene product.

E1 Mechanism for the Dehydration of Alcohols into Alkenes
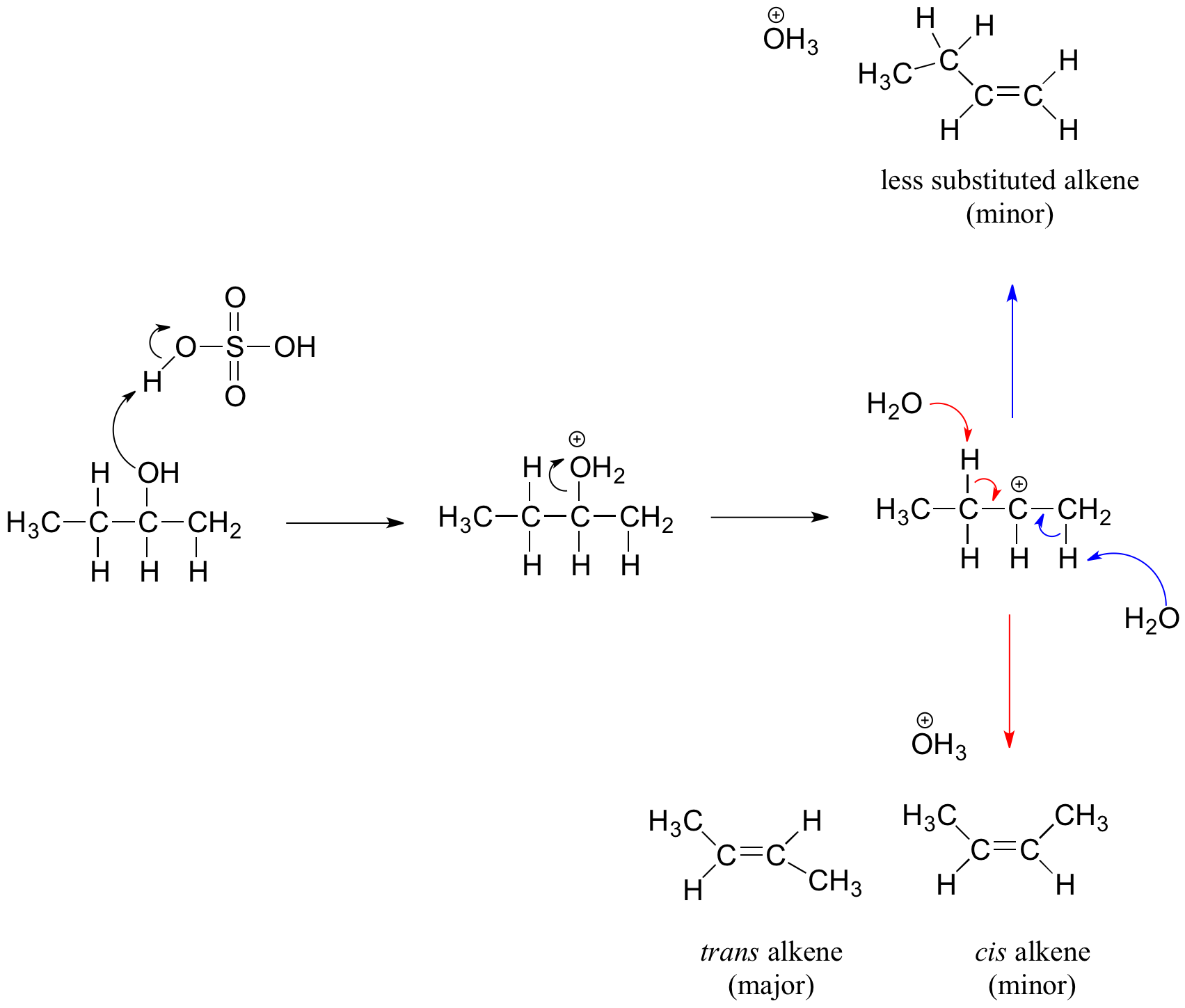
Secondary and tertiary alcohols dehydrate through the E1 mechanism. Similarly to the reaction above, secondary and tertiary –OH protonate to form alkyloxonium ions. However, in this case the ion leaves first to form a carbocation a reaction intermediate. The eliminated water molecule (which is a stronger base than the HSO4- ion) then abstracts a proton from an adjacent carbon, forming a double bond.
Notice in the mechanism below that the alkene formed depends on which adjacent proton is abstracted: the red arrows show formation of the more substituted 2-butene, while the blue arrows show formation of the less substituted 1-butene. This reaction also follows Zaitsev's rule which states that the most stable alkene will be the major product.
As a general rule that more substituted alkenes are more stable than less substituted alkenes. Also, trans alkenes are more stable than cis alkenes. Therefore, the trans isomer of the 2-butene product is expected to be the major product of this reaction.
Similar biological dehydrations are fairly common. These reaction usually occur by an E1cB mechanism where the OH leaving group is two carbons away from a carbonyl group. An example is the dehydration of 5-dehydroquinate to form 5-dehydroshikimate as part of aromatic amino acid biosynthesis of tyrosine. Initially a biological deprotonates the carbon adjacent to the carbonyl group. The resulting carbanion intermediate forms a double bond by eliminating an OH group. This OH group is protonated by an acid and form water.

Conversion of Alcohols into Alkenes Using POCl3
The E2 elimination of 2o and 3º-alcohols under mild, basic conditions may be accomplished by treatment with phosphorous oxychloride (POCl3) in pyridine. Pryidine is typically used as the reaction solvent and acts as the base which removes an adjacent proton in the E2 mechanism.

Mechanism
The reaction starts with the reactant alcohol undergoing a substitution reaction with POCl3 to form a dichlorophospate intermediate. Next pyridine removes an adjacent proton and the dichlorophospate group is eliminated as a leaving group to form a double bond by an E2 mechanism.
1) Formation of a dichlorphosphonium intermediate.

2) Deprotonation of the dichlorophosphonium intermediate to form a dichlorophosphate.

2) E2 elimination

Conversion of Alcohols into Esters
Alcohols can be converted into esters through reaction with carboxylic acids using a strong acid as a catalyst. Often this transformation is performed by increasing the reactivity of the carboxylic acid by first converting it into a carboxylic acid chloride functional group by reacting with thionyl chloride (SOCl2). The acid chloride can then be reacted with an alcohol to create an ester. These reactions will be discussed in greater detail in Chapter 21.



In biological systems, ester formation occurs through a similar process except thioester or acyl adenosyl phosphates are used instead of carboxylic acid chlorides. These reaction will be discussed in more detail in Chapter 21.

Exercises
1) Draw the expected product of the reaction of cylohexanol with the following reagents.
(a) SOCl2 (b) PBr3
2) Draw the expected product of the treatment of the following compounds with OPCl3 in pyridine.
a)  b)
b)  c)
c)  d)
d)  e)
e) 
- Answer
-
1) a) Chlorocyclohexane b) Bromocyclohexane
2) a) 2-Methyl-2-butene b) 3-Methylcyclopentene c) 1-Methylcyclopentene d) 2,3-Dimethyl-2-butene e) 2-Methyl-2-butene
Draw the expected product of the reaction of cylohexanol with the following reagents.
(A) CrO3, H2SO4, H2O (B) Dess-Martin Periodinane (C) SOCl2 (D) NaH and 1-bromoethane (E) PBr3
- Answer
-

Starting with cyclohexanol, describe how you would prepare the following?
(A) cyclohexyl acetate (B) 1-allylcyclohexan-1-ol (C) cyclohexene (D) ethoxycyclohexane
- Answer
-
A) This can be seen as a transesterification, acid and some other ester would be needed to form cyclohexylacetate
B) First, oxidize the alcohol to a ketone, take for example Dess-Martin Periodinane, then use an allyl grignard to form 1-allylcyclohexan-1-ol
C) Alcohols can dehydrate to form alkenes under acidic conditions, so using anhydrous acid and heat would yield cyclohexene
D) The alcohol can also be a nucleophile, perform a halogen substitution, using 1-X ethane, to yield ethoxycyclohexane
In cyclohexanone, a ketone, indicate the polarity of the bond between oxygen and carbon.
- Answer
-

Oxygen is more electronegative than carbon creating the polar bond. This is the basis for the carbon's electrophilicity.
In the dehydration of 1-methylcyclohexanol, which product is favored?

- Answer
-
The more substituted alkene is favored, as more substituted alkenes are relatively lower in energy.
In the dehydration of this diol the resulting product is a ketone. Draw the mechanism of its formation. (Hint: a rearrangement occurs.)

- Answer
-

Draw the mechanism of the reaction of thinoylchloride with cyclohexanol, given below.

- Answer
-

Draw an arrow pushing mechanism for the acid catalyzed dehydration of the following alcohol, make sure to draw both potential mechanisms. Assume no rearrangement for the first two product mechanisms. Which of these two would likely be the major product? If there was a rearrangement, draw the expected major product.

- Answer
-
The major product of this mechanism would be the more highly substituted alkene, or the product formed from the red arrows.

Note the secondary carbocation adjacent a tertiary carbon center, if there were a hydride transfer (rearrangement) to form a tertiary carbocation the following would be the major product. The minor product being the same product as the one formed from the red arrows.

The following epoxide can be transformed into an alcohol using a grignard reagent, take for example allylmagnesium chloride. Draw the product of the treatment of this epoxide with this grignard after being worked up with H2O. Note the stereochemistry and also remember that benzyllic carbons are good SN2 electrophiles.

- Answer
-
Add texts here. Do not delete this text first.

As seen in the previous example, there are many examples of chiral compounds containing alcohols. One common example of these are sugars, is the given the following sugar, allitol, also chiral?

- Answer
-
This compound actually has a plane of symmetry, the plane parallel to the carbon chain/backbone. So, it is not chiral, also called a meso compound.