
12.5: High-Performance Liquid Chromatography
- Page ID
- 152403

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
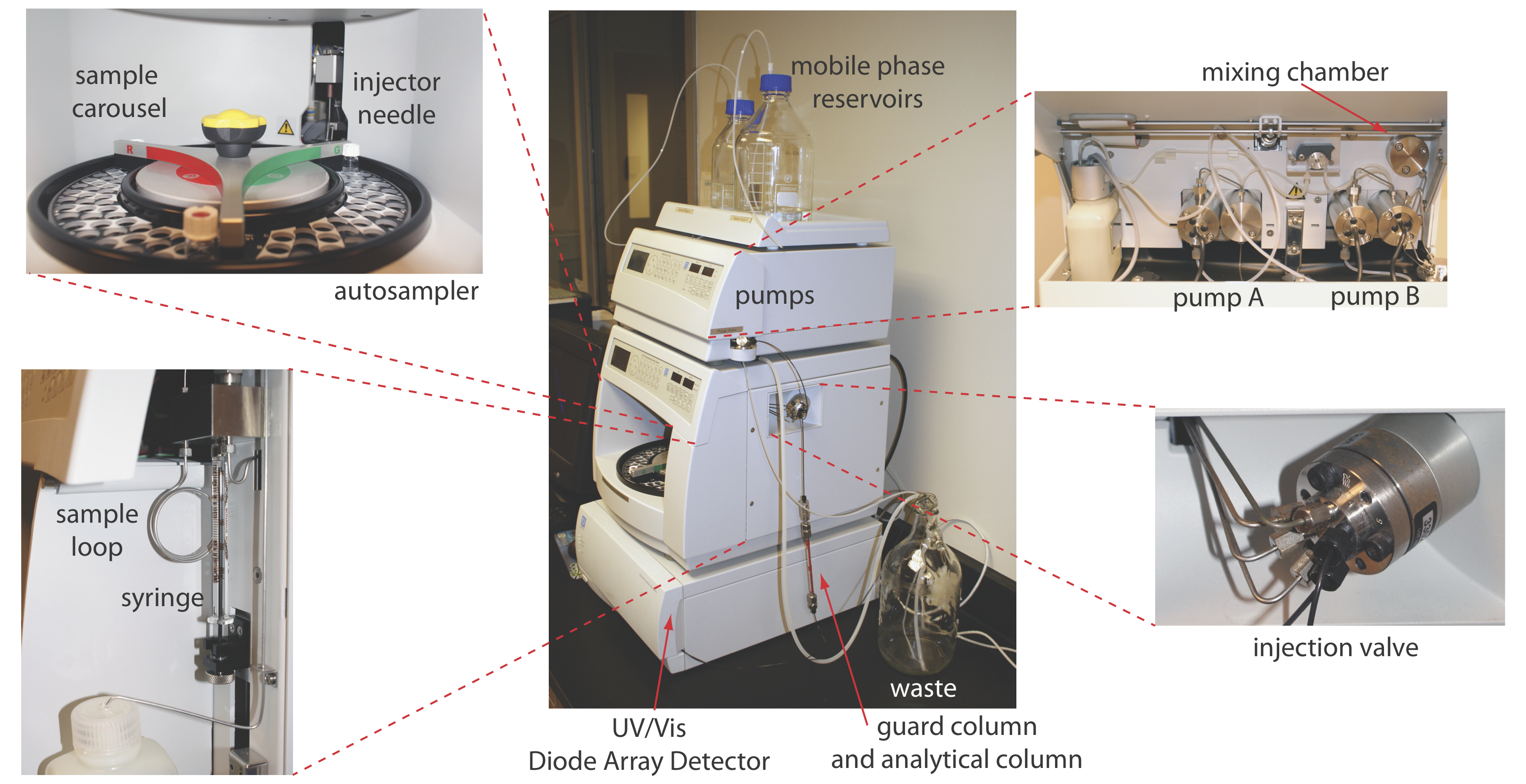
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)In high-performance liquid chromatography (HPLC) we inject the sample, which is in solution form, into a liquid mobile phase. The mobile phase carries the sample through a packed or capillary column that separates the sample’s components based on their ability to partition between the mobile phase and the stationary phase. Figure 12.5.1 shows an example of a typical HPLC instrument, which has several key components: reservoirs that store the mobile phase; a pump for pushing the mobile phase through the system; an injector for introducing the sample; a column for separating the sample into its component parts; and a detector for monitoring the eluent as it comes off the column. Let’s consider each of these components.
A solute’s retention time in HPLC is determined by its interaction with the stationary phase and the mobile phase. There are several different types of solute/stationary phase interactions, including liquid–solid adsorption, liquid–liquid partitioning, ion-exchange, and size-exclusion. This chapter deals exclusively with HPLC separations based on liquid–liquid partitioning. Other forms of liquid chromatography receive consideration in Chapter 12.6.
HPLC Columns
An HPLC typically includes two columns: an analytical column, which is responsible for the separation, and a guard column that is placed before the analytical column to protect it from contamination.
Analytical Columns
The most common type of HPLC column is a stainless steel tube with an internal diameter between 2.1 mm and 4.6 mm and a length between 30 mm and 300 mm (Figure 12.5.2 ). The column is packed with 3–10 µm porous silica particles with either an irregular or a spherical shape. Typical column efficiencies are 40000–60000 theoretical plates/m. Assuming a Vmax/Vmin of approximately 50, a 25-cm column with 50 000 plates/m has 12 500 theoretical plates and a peak capacity of 110.

Capillary columns use less solvent and, because the sample is diluted to a lesser extent, produce larger signals at the detector. These columns are made from fused silica capillaries with internal diameters from 44–200 μm and lengths of 50–250 mm. Capillary columns packed with 3–5 μm particles have been prepared with column efficiencies of up to 250 000 theoretical plates [Novotony, M. Science, 1989, 246, 51–57].
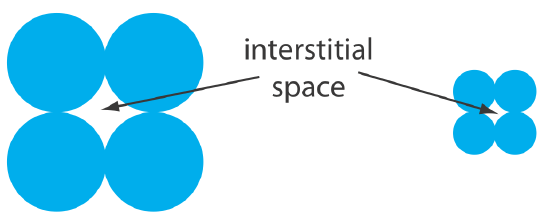
One limitation to a packed capillary column is the back pressure that develops when pumping the mobile phase through the small interstitial spaces between the particulate micron-sized packing material (Figure 12.5.3 ). Because the tubing and fittings that carry the mobile phase have pressure limits, a higher back pressure requires a lower flow rate and a longer analysis time. Monolithic columns, in which the solid support is a single, porous rod, offer column efficiencies equivalent to a packed capillary column while allowing for faster flow rates. A monolithic column—which usually is similar in size to a conventional packed column, although smaller, capillary columns also are available—is prepared by forming the mono- lithic rod in a mold and covering it with PTFE tubing or a polymer resin. Monolithic rods made of a silica-gel polymer typically have macropores with diameters of approximately 2 μm and mesopores—pores within the macropores—with diameters of approximately 13 nm [Cabrera, K. Chromatography Online, April 1, 2008].
Guard Columns
Two problems tend to shorten the lifetime of an analytical column. First, solutes that bind irreversibly to the stationary phase degrade the column’s performance by decreasing the amount of stationary phase available for effecting a separation. Second, particulate material injected with the sample may clog the analytical column. To minimize these problems we place a guard column before the analytical column. A Guard column usually contains the same particulate packing material and stationary phase as the analytical column, but is significantly shorter and less expensive—a length of 7.5 mm and a cost one-tenth of that for the corresponding analytical column is typical. Because they are intended to be sacrificial, guard columns are replaced regularly.
If you look closely at Figure 12.5.1 , you will see the small guard column just above the analytical column.
Stationary Phases for Liquid-Liquid Chromatography
In liquid–liquid chromatography the stationary phase is a liquid film coated on a packing material, typically 3–10 μm porous silica particles. Because the stationary phase may be partially soluble in the mobile phase, it may elute, or bleed from the column over time. To prevent the loss of stationary phase, which shortens the column’s lifetime, it is bound covalently to the silica particles. Bonded stationary phases are created by reacting the silica particles with an organochlorosilane of the general form Si(CH3)2RCl, where R is an alkyl or substituted alkyl group.

To prevent unwanted interactions between the solutes and any remaining –SiOH groups, Si(CH3)3Cl is used to convert unreacted sites to \(–\text{SiOSi(CH}_3)_3\); such columns are designated as end-capped.
The properties of a stationary phase depend on the organosilane’s alkyl group. If R is a polar functional group, then the stationary phase is polar. Examples of polar stationary phases include those where R contains a cyano (–C2H4CN), a diol (–C3H6OCH2CHOHCH2OH), or an amino (–C3H6NH2) functional group. Because the stationary phase is polar, the mobile phase is a nonpolar or a moderately polar solvent. The combination of a polar stationary phase and a nonpolar mobile phase is called normal- phase chromatography.
In reversed-phase chromatography, which is the more common form of HPLC, the stationary phase is nonpolar and the mobile phase is polar. The most common nonpolar stationary phases use an organochlorosilane where the R group is an n-octyl (C8) or n-octyldecyl (C18) hydrocarbon chain. Most reversed-phase separations are carried out using a buffered aqueous solution as a polar mobile phase, or using other polar solvents, such as methanol and acetonitrile. Because the silica substrate may undergo hydrolysis in basic solutions, the pH of the mobile phase must be less than 7.5.
It seems odd that the more common form of liquid chromatography is identified as reverse-phase instead of normal phase. You might recall that one of the earliest examples of chromatography was Mikhail Tswett’s separation of plant pigments using a polar column of calcium carbonate and a nonpolar mobile phase of petroleum ether. The assignment of normal and reversed, therefore, is all about precedence.
Mobile Phases
The elution order of solutes in HPLC is governed by polarity. For a normal-phase separation, a solute of lower polarity spends proportionally less time in the polar stationary phase and elutes before a solute that is more polar. Given a particular stationary phase, retention times in normal-phase HPLC are controlled by adjusting the mobile phase’s properties. For example, if the resolution between two solutes is poor, switching to a less polar mobile phase keeps the solutes on the column for a longer time and provides more opportunity for their separation. In reversed-phase HPLC the order of elution is the opposite that in a normal-phase separation, with more polar solutes eluting first. Increasing the polarity of the mobile phase leads to longer retention times. Shorter retention times require a mobile phase of lower polarity.
Choosing a Mobile Phase: Using the Polarity Index
There are several indices that help in selecting a mobile phase, one of which is the polarity index [Snyder, L. R.; Glajch, J. L.; Kirkland, J. J. Practical HPLC Method Development, Wiley-Inter- science: New York, 1988]. Table 12.5.1 provides values of the polarity index, \(P^{\prime}\), for several common mobile phases, where larger values of \(P^{\prime}\) correspond to more polar solvents. Mixing together two or more mobile phases—assuming they are miscible—creates a mobile phase of intermediate polarity. For example, a binary mobile phase made by combining solvent A and solvent B has a polarity index, \(P_{AB}^{\prime}\), of
\[P_{A B}^{\prime}=\Phi_{A} P_{A}^{\prime}+\Phi_{B} P_{B}^{\prime} \label{12.1}\]
where \(P_A^{\prime}\) and \(P_B^{\prime}\) are the polarity indices for solvents A and B, and \(\Phi_A\) and \(\Phi_B\) are the volume fractions for the two solvents.
A reversed-phase HPLC separation is carried out using a mobile phase of 60% v/v water and 40% v/v methanol. What is the mobile phase’s polarity index?
Solution
Using Equation \ref{12.1} and the values in Table 12.5.1 , the polarity index for a 60:40 water–methanol mixture is
\[P_{A B}^{\prime}=\Phi_\text{water} P_\text{water}^{\prime}+\Phi_\text{methanol} P_\text{methanol}^{\prime} \nonumber\]
\[P_{A B}^{\prime}=0.60 \times 10.2+0.40 \times 5.1=8.2 \nonumber\]
Suppose you need a mobile phase with a polarity index of 7.5. Explain how you can prepare this mobile phase using methanol and water.
- Answer
-
If we let x be the fraction of water in the mobile phase, then 1 – x is the fraction of methanol. Substituting these values into Equation \ref{12.1} and solving for x
\[7.5=10.2 x+5.1(1-x) \nonumber\]
\[7.5=10.2 x+5.1-5.1 x \nonumber\]
\[2.4=5.1 x \nonumber\]
gives x as 0.47. The mobile phase is 47% v/v water and 53% v/v methanol.
As a general rule, a two unit change in the polarity index corresponds to an approximately 10-fold change in a solute’s retention factor. Here is a simple example. If a solute’s retention factor, k, is 22 when using water as a mobile phase (\(P^{\prime}\) = 10.2), then switching to a mobile phase of 60:40 water–methanol (\(P^{\prime}\) = 8.2) decreases k to approximately 2.2. Note that the retention factor becomes smaller because we are switching from a more polar mobile phase to a less polar mobile phase in a reversed-phase separation.
Choosing a Mobile Phase: Adjusting Selectivity
Changing the mobile phase’s polarity index changes a solute’s retention factor. As we learned in Chapter 12.3, however, a change in k is not an effective way to improve resolution when the initial value of k is greater than 10. To effect a better separation between two solutes we must improve the selectivity factor, \(\alpha\). There are two common methods for increasing \(\alpha\): adding a reagent to the mobile phase that reacts with the solutes in a secondary equilibrium reaction or switching to a different mobile phase.
Taking advantage of a secondary equilibrium reaction is a useful strategy for improving a separation [(a) Foley, J. P. Chromatography, 1987, 7, 118–128; (b) Foley, J. P.; May, W. E. Anal. Chem. 1987, 59, 102–109; (c) Foley, J. P.; May, W. E. Anal. Chem. 1987, 59, 110–115]. Figure 12.3.3, which we considered earlier in this chapter, shows the reversed-phase separation of four weak acids—benzoic acid, terephthalic acid, p-aminobenzoic acid, and p-hydroxybenzoic acid—on a nonpolar C18 column using an aqueous buffer of acetic acid and sodium acetate as the mobile phase. The retention times for these weak acids are shorter when using a less acidic mobile phase because each solute is present in an anionic, weak base form that is less soluble in the nonpolar stationary phase. If the mobile phase’s pH is sufficiently acidic, the solutes are present as neutral weak acids that are more soluble in the stationary phase and take longer to elute. Because the weak acid solutes do not have identical pKa values, the pH of the mobile phase has a different effect on each solute’s retention time, allowing us to find the optimum pH for effecting a complete separation of the four solutes.
Acid–base chemistry is not the only example of a secondary equilibrium reaction. Other examples include ion-pairing, complexation, and the interaction of solutes with micelles. We will consider the last of these in Chapter 12.7 when we discuss micellar electrokinetic capillary chromatography.
In Example 12.5.1 we learned how to adjust the mobile phase’s polarity by blending together two solvents. A polarity index, however, is just a guide, and binary mobile phase mixtures with identical polarity indices may not resolve equally a pair of solutes. Table 12.5.2 , for example, shows retention times for four weak acids in two mobile phases with nearly identical values for \(P^{\prime}\). Although the order of elution is the same for both mobile phases, each solute’s retention time is affected differently by the choice of organic solvent. If we switch from using acetonitrile to tetrahydrofuran, for example, we find that benzoic acid elutes more quickly and that p-hydroxybenzoic acid elutes more slowly. Although we can resolve fully these two solutes using mobile phase that is 16% v/v acetonitrile, we cannot resolve them if the mobile phase is 10% tetrahydrofuran.
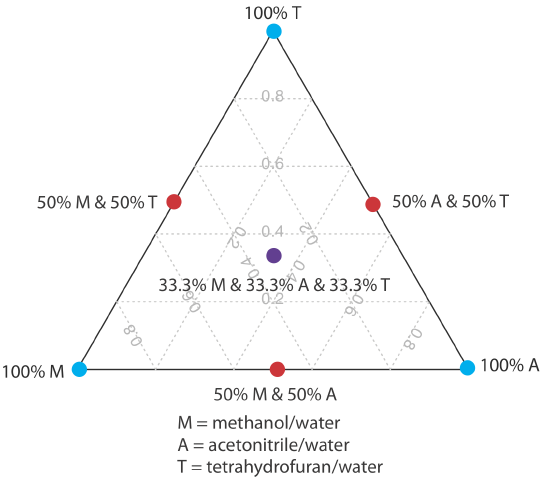
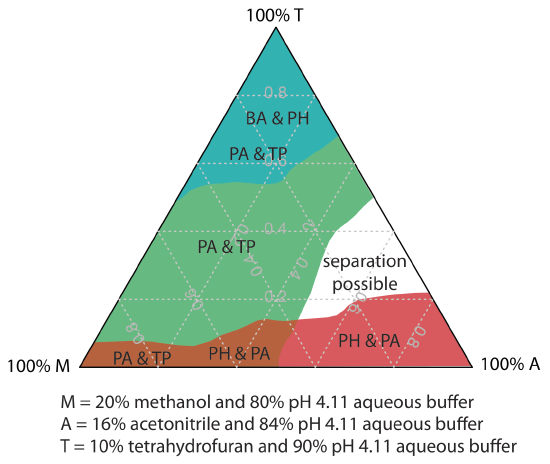
One strategy for finding the best mobile phase is to use the solvent triangle shown in Figure 12.5.4 , which allows us to explore a broad range of mobile phases with only seven experiments. We begin by adjusting the amount of acetonitrile in the mobile phase to produce the best possible separation within the desired analysis time. Next, we use Table 12.5.3 to estimate the composition of methanol/H2O and tetrahydrofuran/H2O mobile phases that will produce similar analysis times. Four additional mobile phases are prepared using the binary and ternary mobile phases shown in Figure 12.5.4 . When we examine the chromatograms from these seven mobile phases we may find that one or more provides an adequate separation, or we may identify a region within the solvent triangle where a separation is feasible. Figure 12.5.5 shows a resolution map for the reversed-phase separation of benzoic acid, terephthalic acid, p-aminobenzoic acid, and p-hydroxybenzoic acid on a nonpolar C18 column in which the maximum desired analysis time is set to 6 min [Harvey, D. T.; Byerly, S.; Bowman, A.; Tomlin, J. J. Chem. Educ. 1991, 68, 162–168]. The areas in blue, green, and red show mobile phase compositions that do not provide baseline resolution. The unshaded area represents mobile phase compositions where a separation is possible.
The choice to start with acetonitrile is arbitrary—we can just as easily choose to begin with methanol or with tetrahydrofuran.
Choosing a Mobile Phase: Isocratic and Gradient Elutions
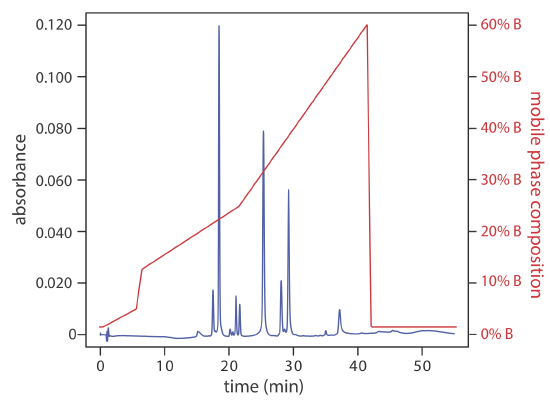
A separation using a mobile phase that has a fixed composition is an isocratic elution. One difficulty with an isocratic elution is that an appropriate mobile phase strength for resolving early-eluting solutes may lead to unacceptably long retention times for late-eluting solutes. Optimizing the mobile phase for late-eluting solutes, on the other hand, may provide an inadequate separation of early-eluting solutes. Changing the mobile phase’s composition as the separation progresses is one solution to this problem. For a reversed-phase separation we use an initial mobile phase that is more polar. As the separation progresses, we adjust the composition of mobile phase so that it becomes less polar (see Figure 12.5.6 ). Such separations are called gradient elutions.
HPLC Plumbing
In a gas chromatograph the pressure from a compressed gas cylinder is sufficient to push the mobile phase through the column. Pushing a liquid mobile phase through a column, however, takes a great deal more effort, generating pressures in excess of several hundred atmospheres. In this section we consider the basic plumbing needed to move the mobile phase through the column and to inject the sample into the mobile phase.
Moving the Mobile Phase
A typical HPLC includes between 1–4 reservoirs for storing mobile phase solvents. The instrument in Figure 12.5.1 , for example, has two mobile phase reservoirs that are used for an isocratic elution or a gradient elution by drawing solvents from one or both reservoirs.
Before using a mobile phase solvent we must remove dissolved gases, such as N2 and O2, and small particulate matter, such as dust. Because there is a large drop in pressure across the column—the pressure at the column’s entrance is as much as several hundred atmospheres, but it is atmospheric pressure at the column’s exit—gases dissolved in the mobile phase are released as gas bubbles that may interfere with the detector’s response. Degassing is accomplished in several ways, but the most common are the use of a vacuum pump or sparging with an inert gas, such as He, which has a low solubility in the mobile phase. Particulate materials, which may clog the HPLC tubing or column, are removed by filtering the solvents.
Bubbling an inert gas through the mobile phase releases volatile dissolved gases. This process is called sparging.
The mobile phase solvents are pulled from their reservoirs by the action of one or more pumps. Figure 12.5.7 shows a close-up view of the pumps for the instrument in Figure 12.5.1 . The working pump and the equilibrating pump each have a piston whose back and forth movement maintains a constant flow rate of up to several mL/min and provides the high output pressure needed to push the mobile phase through the chromatographic column. In this particular instrument, each pump sends its mobile phase to a mixing chamber where they combine to form the final mobile phase. The relative speed of the two pumps determines the mobile phase’s final composition.
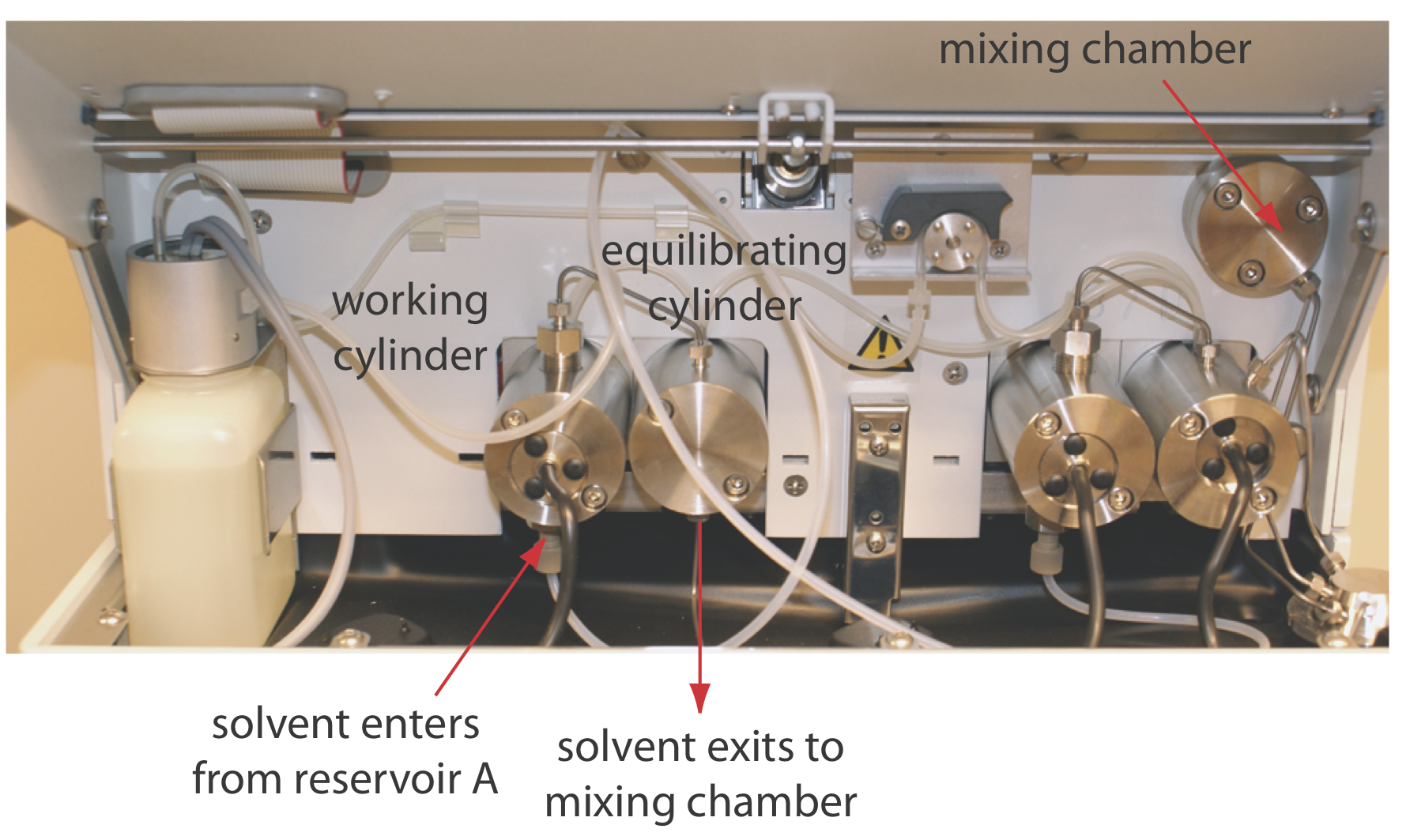
The back and forth movement of a reciprocating pump creates a pulsed flow that contributes noise to the chromatogram. To minimize these pulses, each pump in Figure 12.5.7 has two cylinders. During the working cylinder’s forward stoke it fills the equilibrating cylinder and establishes flow through the column. When the working cylinder is on its reverse stroke, the flow is maintained by the piston in the equilibrating cylinder. The result is a pulse-free flow.
There are other possible ways to control the mobile phase’s composition and flow rate. For example, instead of the two pumps in Figure 12.5.7 , we can place a solvent proportioning valve before a single pump. The solvent proportioning value connects two or more solvent reservoirs to the pump and determines how much of each solvent is pulled during each of the pump’s cycles. Another approach for eliminating a pulsed flow is to include a pulse damper between the pump and the column. A pulse damper is a chamber filled with an easily compressed fluid and a flexible diaphragm. During the piston’s forward stroke the fluid in the pulse damper is compressed. When the piston withdraws to refill the pump, pressure from the expanding fluid in the pulse damper maintains the flow rate.
Injecting the Sample
The operating pressure within an HPLC is sufficiently high that we cannot inject the sample into the mobile phase by inserting a syringe through a septum, as is possible in gas chromatography. Instead, we inject the sample using a loop injector, a diagram of which is shown in Figure 12.5.8 . In the load position a sample loop—which is available in a variety of sizes ranging from 0.5 μL to 5 mL—is isolated from the mobile phase and open to the atmosphere. The sample loop is filled using a syringe with a capacity several times that of the sample loop, with excess sample exiting through the waste line. After loading the sample, the injector is turned to the inject position, which redirects the mobile phase through the sample loop and onto the column.
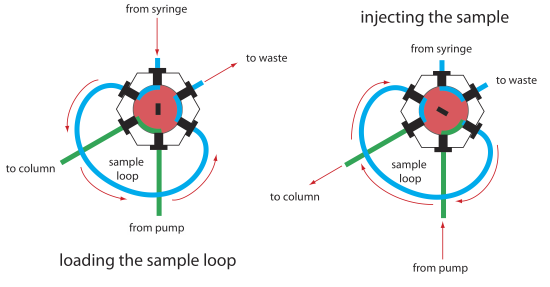
The instrument in Figure 12.5.1 uses an autosampler to inject samples. Instead of using a syringe to push the sample into the sample loop, the syringe draws sample into the sample loop.
Detectors for HPLC
Many different types of detectors have been use to monitor HPLC separations, most of which use the spectroscopic techniques from Chapter 10 or the electrochemical techniques from Chapter 11.
Spectroscopic Detectors
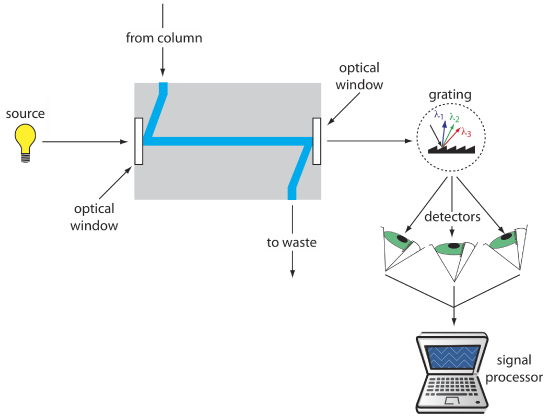
The most popular HPLC detectors take advantage of an analyte’s UV/Vis absorption spectrum. These detectors range from simple designs, in which the analytical wavelength is selected using appropriate filters, to a modified spectrophotometer in which the sample compartment includes a flow cell. Figure 12.5.9 shows the design of a typical flow cell when using a diode array spectrometer as the detector. The flow cell has a volume of 1–10 μL and a path length of 0.2–1 cm.
To review the details of how we measure absorbance, see Chapter 10.2. More information about different types of instruments, including the diode array spectrometer, is in Chapter 10.3.
When using a UV/Vis detector the resulting chromatogram is a plot of absorbance as a function of elution time (see Figure 12.5.10 ). If the detector is a diode array spectrometer, then we also can display the result as a three-dimensional chromatogram that shows absorbance as a function of wavelength and elution time. One limitation to using absorbance is that the mobile phase cannot absorb at the wavelengths we wish to monitor. Table 12.5.1 lists the minimum useful UV wavelength for several common HPLC solvents. Absorbance detectors provide detection limits of as little as 100 pg–1 ng of injected analyte.
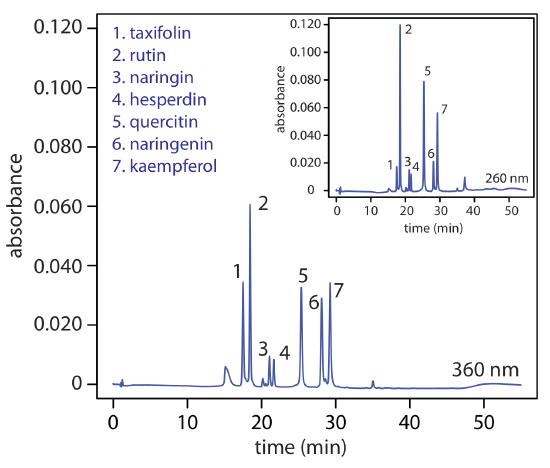
If an analyte is fluorescent, we can place the flow cell in a spectrofluorimeter. As shown in Figure 12.5.11 , a fluorescence detector provides additional selectivity because only a few of a sample’s components are fluorescent. Detection limits are as little as 1–10 pg of injected analyte.
See Chapter 10.6 for a review of fluorescence spectroscopy and spectrofluorimeters.
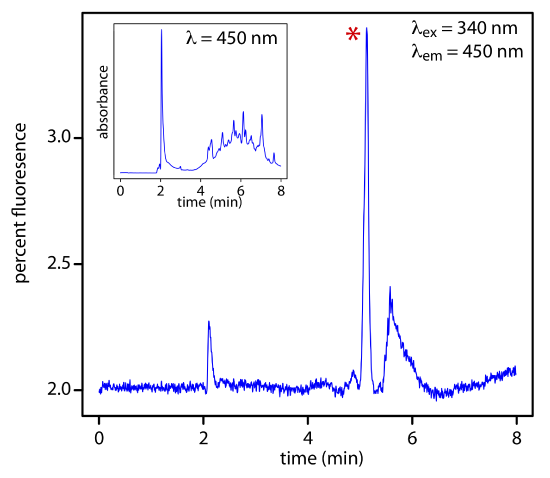
Electrochemical Detectors
Another common group of HPLC detectors are those based on electrochemical measurements such as amperometry, voltammetry, coulometry, and conductivity. Figure 12.5.12 , for example, shows an amperometric flow cell. Effluent from the column passes over the working electrode—held at a constant potential relative to a downstream reference electrode—that completely oxidizes or reduces the analytes. The current flowing between the working electrode and the auxiliary electrode serves as the analytical signal. Detection limits for amperometric electrochemical detection are from 10 pg–1 ng of injected analyte.
See Chapter 11.4 for a review of amperometry.
Other Detectors
Several other detectors have been used in HPLC. Measuring a change in the mobile phase’s refractive index is analogous to monitoring the mobile phase’s thermal conductivity in gas chromatography. A refractive index detector is nearly universal, responding to almost all compounds, but has a relatively poor detection limit of 0.1–1 μg of injected analyte. An additional limitation of a refractive index detector is that it cannot be used for a gradient elution unless the mobile phase components have identical refractive indexes.
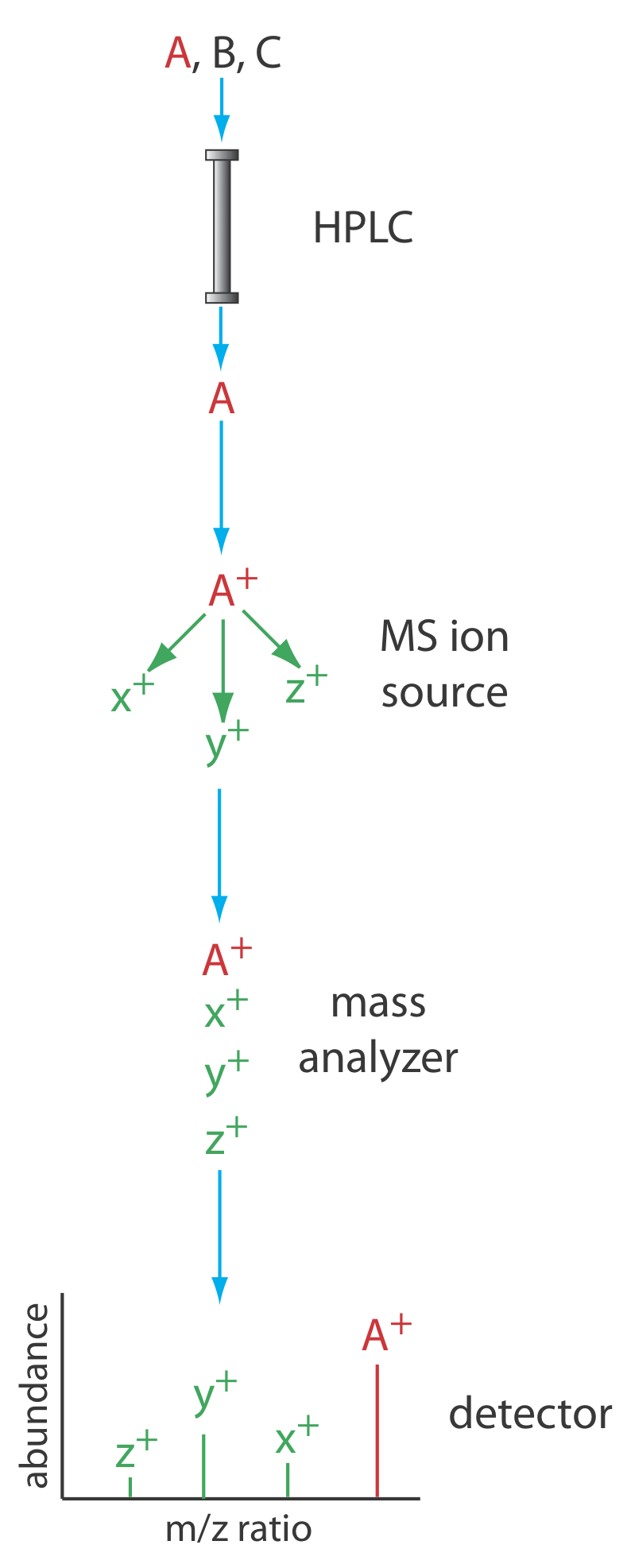
Another useful detector is a mass spectrometer. Figure 12.5.13 shows a block diagram of a typical HPLC–MS instrument. The effluent from the column enters the mass spectrometer’s ion source using an interface the removes most of the mobile phase, an essential need because of the incompatibility between the liquid mobile phase and the mass spectrometer’s high vacuum environment. In the ionization chamber the remaining molecules—a mixture of the mobile phase components and solutes—undergo ionization and fragmentation. The mass spectrometer’s mass analyzer separates the ions by their mass-to-charge ratio (m/z). A detector counts the ions and displays the mass spectrum.
There are several options for monitoring the chromatogram when using a mass spectrometer as the detector. The most common method is to continuously scan the entire mass spectrum and report the total signal for all ions reaching the detector during each scan. This total ion scan provides universal detection for all analytes. As seen in Figure 12.5.14 , we can achieve some degree of selectivity by monitoring only specific mass-to-charge ratios, a process called selective-ion monitoring.
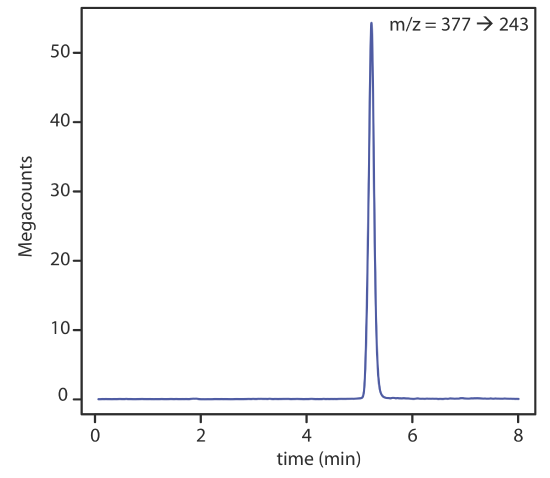
The advantages of using a mass spectrometer in HPLC are the same as for gas chromatography. Detection limits are very good, typically 0.1–1 ng of injected analyte, with values as low as 1–10 pg for some samples. In addition, a mass spectrometer provides qualitative, structural information that can help to identify the analytes. The interface between the HPLC and the mass spectrometer is technically more difficult than that in a GC–MS because of the incompatibility of a liquid mobile phase with the mass spectrometer’s high vacuum requirement.
For more details on mass spectrometry see Introduction to Mass Spectrometry by Michael Samide and Olujide Akinbo, a resource that is part of the Analytical Sciences Digital Library.
Quantitative Applications
High-performance liquid chromatography is used routinely for both qualitative and quantitative analyses of environmental, pharmaceutical, industrial, forensic, clinical, and consumer product samples.
Preparing Samples for Analysis
Samples in liquid form are injected into the HPLC after a suitable clean-up to remove any particulate materials, or after a suitable extraction to remove matrix interferents. In determining polyaromatic hydrocarbons (PAH) in wastewater, for example, an extraction with CH2Cl2 serves the dual purpose of concentrating the analytes and isolating them from matrix interferents. Solid samples are first dissolved in a suitable solvent or the analytes of interest brought into solution by extraction. For example, an HPLC analysis for the active ingredients and the degradation products in a pharmaceutical tablet often begins by extracting the powdered tablet with a portion of mobile phase. Gas samples are collected by bubbling them through a trap that contains a suitable solvent. Organic isocyanates in industrial atmospheres are collected by bubbling the air through a solution of 1-(2-methoxyphenyl)piperazine in toluene. The reaction between the isocyanates and 1-(2-methoxyphenyl)piperazine both stabilizes them against degradation before the HPLC analysis and converts them to a chemical form that can be monitored by UV absorption.
Quantitative Calculations
A quantitative HPLC analysis is often easier than a quantitative GC analysis because a fixed volume sample loop provides a more precise and accurate injection. As a result, most quantitative HPLC methods do not need an internal standard and, instead, use external standards and a normal calibration curve.
An internal standard is necessary when using HPLC–MS because the interface between the HPLC and the mass spectrometer does not allow for a reproducible transfer of the column’s eluent into the MS’s ionization chamber.
The concentration of polynuclear aromatic hydrocarbons (PAH) in soil is determined by first extracting the PAHs with methylene chloride. The extract is diluted, if necessary, and the PAHs separated by HPLC using a UV/Vis or fluorescence detector. Calibration is achieved using one or more external standards. In a typical analysis a 2.013-g sample of dried soil is extracted with 20.00 mL of methylene chloride. After filtering to remove the soil, a 1.00-mL portion of the extract is removed and diluted to 10.00 mL with acetonitrile. Injecting 5 μL of the diluted extract into an HPLC gives a signal of 0.217 (arbitrary units) for the PAH fluoranthene. When 5 μL of a 20.0-ppm fluoranthene standard is analyzed using the same conditions, a signal of 0.258 is measured. Report the parts per million of fluoranthene in the soil.
Solution
For a single-point external standard, the relationship between the signal, S, and the concentration, C, of fluoranthene is
\[S = kC \nonumber\]
Substituting in values for the standard’s signal and concentration gives the value of k as
\[k=\frac{S}{C}=\frac{0.258}{20.0 \text{ ppm}}=0.0129 \text{ ppm}^{-1} \nonumber\]
Using this value for k and the sample’s HPLC signal gives a fluoranthene concentration of
\[C=\frac{S}{k}=\frac{0.217}{0.0129 \text{ ppm}^{-1}}=16.8 \text{ ppm} \nonumber\]
for the extracted and diluted soil sample. The concentration of fluoranthene in the soil is
\[\frac{16.8 \text{ g} / \mathrm{mL} \times \frac{10.00 \text{ mL}}{1.00 \text{ mL}} \times 20.00 \text{ mL}}{2.013 \text{ g} \text { sample }}=1670 \text{ ppm} \text { fluoranthene } \nonumber\]
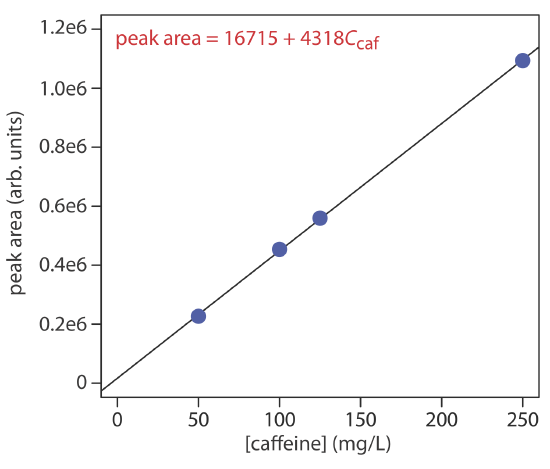
The concentration of caffeine in beverages is determined by a reversed-phase HPLC separation using a mobile phase of 20% acetonitrile and 80% water, and using a nonpolar C8 column. Results for a series of 10-μL injections of caffeine standards are in the following table.
| [caffeine] (mg/L) | peak area (arb. units) |
|---|---|
| 50.0 | 226724 |
| 100.0 | 453762 |
| 125.0 | 559443 |
| 250.0 | 1093637 |
What is the concentration of caffeine in a sample if a 10-μL injection gives a peak area of 424195? The data in this problem comes from Kusch, P.; Knupp, G. “Simultaneous Determination of Caffeine in Cola Drinks and Other Beverages by Reversed-Phase HPTLC and Reversed-Phase HPLC,” Chem. Educator, 2003, 8, 201–205.
- Answer
-
The figure below shows the calibration curve and calibration equation for the set of external standards. Substituting the sample’s peak area into the calibration equation gives the concentration of caffeine in the sample as 94.4 mg/L.
The best way to appreciate the theoretical and the practical details discussed in this section is to carefully examine a typical analytical method. Although each method is unique, the following description of the determination of fluoxetine in serum provides an instructive example of a typical procedure. The description here is based on Smyth, W. F. Analytical Chemistry of Complex Matricies, Wiley Teubner: Chichester, England, 1996, pp. 187–189.
Representative Method 12.5.1: Determination of Fluoxetine in Serum
Description of Method
Fluoxetine is another name for the antidepressant drug Prozac. The determination of fluoxetine in serum is an important part of monitoring its therapeutic use. The analysis is complicated by the complex matrix of serum samples. A solid-phase extraction followed by an HPLC analysis using a fluorescence detector provides the necessary selectivity and detection limits.
Procedure
Add a known amount of the antidepressant protriptyline, which serves as an internal standard, to each serum sample and to each external standard. To remove matrix interferents, pass a 0.5-mL aliquot of each serum sample or standard through a C18 solid-phase extraction cartridge. After washing the cartridge to remove the interferents, elute the remaining constituents, including the analyte and the internal standard, by washing the cartridge with 0.25 mL of a 25:75 v/v mixture of 0.1 M HClO4 and acetonitrile. Inject a 20-μL aliquot onto a 15-cm \(\times\) 4.6-mm column packed with a 5 μm C8-bonded stationary phase. The isocratic mobile phase is 37.5:62.5 v/v acetonitrile and water (that contains 1.5 g of tetramethylammonium perchlorate and 0.1 mL of 70% v/v HClO4). Monitor the chromatogram using a fluorescence detector set to an excitation wave- length of 235 nm and an emission wavelength of 310 nm.
Questions
1. The solid-phase extraction is important because it removes constitutions in the serum that might interfere with the analysis. What types of interferences are possible?
Blood serum, which is a complex mixture of compounds, is approximately 92% water, 6–8% soluble proteins, and less than 1% each of various salts, lipids, and glucose. A direct injection of serum is not advisable for three reasons. First, any particulate materials in the serum will clog the column and restrict the flow of mobile phase. Second, some of the compounds in the serum may absorb too strongly to the stationary phase, degrading the column’s performance. Finally, although an HPLC can separate and analyze complex mixtures, an analysis is difficult if the number of constituents exceeds the column’s peak capacity.
2. One advantage of an HPLC analysis is that a loop injector often eliminates the need for an internal standard. Why is an internal standard used in this analysis? What assumption(s) must we make when using the internal standard?
An internal standard is necessary because of uncertainties introduced during the solid-phase extraction. For example, the volume of serum transferred to the solid-phase extraction cartridge, 0.5 mL, and the volume of solvent used to remove the analyte and internal standard, 0.25 mL, are very small. The precision and accuracy with which we can measure these volumes is not as good as when we use larger volumes. For example, if we extract the analyte into a volume of 0.24 mL instead of a volume of 0.25 mL, then the analyte’s concentration increases by slightly more than 4%. In addition, the concentration of eluted analytes may vary from trial-to-trial due to variations in the amount of solution held up by the cartridge. Using an internal standard compensates for these variation. To be useful we must assume that the analyte and the internal standard are retained completely during the initial loading, that they are not lost when the cartridge is washed, and that they are extracted completely during the final elution.
3. Why does the procedure monitor fluorescence instead of monitoring UV absorption?
Fluorescence is a more selective technique for detecting analytes. Many other commonly prescribed antidepressants (and their metabolites) elute with retention times similar to that of fluoxetine. These compounds, however, either do not fluoresce or are only weakly fluorescent.
4. If the peaks for fluoxetine and protriptyline are resolved insufficiently, how might you alter the mobile phase to improve their separation?
Decreasing the amount of acetonitrile and increasing the amount of water in the mobile will increase retention times, providing more time to effect a separation.
Evaluation
With a few exceptions, the scale of operation, accuracy, precision, sensitivity, selectivity, analysis time, and cost for an HPLC method are similar to GC methods. Injection volumes for an HPLC method usually are larger than for a GC method because HPLC columns have a greater capacity. Because it uses a loop injection, the precision of an HPLC method often is better than a GC method. HPLC is not limited to volatile analytes, which means we can analyze a broader range of compounds. Capillary GC columns, on the other hand, have more theoretical plates, and can separate more complex mixtures.