
19.8: Metal Alkyls
- Page ID
- 283224

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Part 1:
With this post we finally reach the defining ligands of organometallic chemistry, alkyls. Metal alkyls feature a metal-carbon σ bond and are usually actor ligands, although some alkyl ligands behave as spectators. Our aim will be to understand the general dependence of the behavior of alkyl ligands on the metal center and the ligand’s substituents. Using this knowledge, we can make meaningful comparisons between related metal alkyl complexes and educated predictions about their likely behavior. Because alkyl ligands are central to organometallic chemistry, I’ve decided to spread this discussion across multiple posts. We’ll deal first with the general properties of metal alkyls.
In the Simplifying the Organometallic Complex series, we decomposed the M–C bond into a positively charged metal and negatively charged carbon. This deconstruction procedure is consistent with the relative electronegativities of carbon and the transition metals. It can be very useful for us to imagine metal alkyls essentially as stabilized carbanions—but it’s also important to understand that M–C bonds run the gamut from extremely ionic and salt-like (NaCH3) to essentially covalent ([HgCH3]+). The reactivity of the alkyl ligand is inversely related to the electronegativity of the metal center.
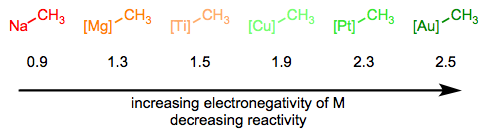
The hybridization of the carbon atom is also important, and the trend here follows the trend in nucleophilicity as a function of hybridization in organic chemistry. sp-Hybridized ligands are the least nucleophilic, followed by sp2 and sp3 ligands respectively.
The history of transition metal alkyls is an intriguing example of an incorrect scientific paradigm. After several unsuccessful attempts to isolate stable metal alkyls, organometallic chemists in the 1920s got the idea that metal-carbon bonds were weak in general. However, later studies showed that it was kinetic instability, not thermodynamic, that was to blame for our inability to isolate metal alkyls. In other words, most metal alkyls are susceptible to decomposition pathways with low activation barriers—the instability of the M–C bond per se is not to blame. Crabtree cites typical values of 30-65 kcal/mol for M–C bond strengths.
What are the major decomposition pathways of metal alkyl complexes? β-hydride elimination is the most common. Thermodynamically, the ubiquity of β-hydride elimination makes sense—M–C bonds run 30-65 kcal/mol, while M–H bonds tend to be stronger. The figure below summarizes the accepted mechanism and requirements of β-hydride elimination. We’ll revisit this fundamental reaction of organometallic complexes in a future post.
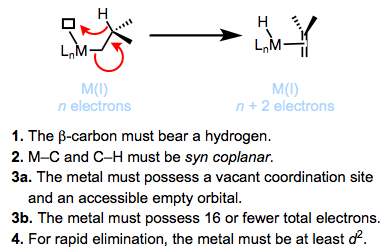
Kinetically stable metal alkyl complexes violate one of the requirements for β-hydride elimination. Methyl and neopentyl complexes lack β-hydrogens, violating requirement 1. Tightly binding, chelating ligands may be used to prevent the formation of an empty coordination site, violating requirements 3a and 3b. Titanium complexes are known that violate requirement 4 and eliminate only very slowly—back-donation from the metal to the σ*C–H is required for rapid elimination (see below).
Reductive elimination is a second common decomposition pathway. The alkyl ligand hooks up with a second X-type ligand on the metal, and the metal is reduced by two units with a decrease in the total electron count by two units. I’ve omitted curved arrows here because different mechanisms of reductive elimination are known. We’ll discuss the requirements of reductive elimination in detail in a future post; for now, it’s important to note that the thermodynamic stability of C–X versus that of (M–X + M–C) is a critical driving force for the reaction.
When X = H, reductive elimination is nearly always thermodynamically favorable; thus, isolable alkyl hydride complexes are rare. This behavior is a feature, not a bug, when we consider that hydrogenation chemistry depends on it! On the other hand, when X = halogen reductive elimination is usually disfavored. Reductive elimination of C–C (X = C) can be favored thermodynamically, but is usually slower than the corresponding C–H elimination.
Complexes that cannot undergo β-hydride elimination are sometimes susceptible to α-elimination, a mechanistically similar process that forms a metal carbene. This process is particularly facile when the α-position is activated by an adjacent electron donor (Fischer carbenes are the result).
In some metal alkyl complexes, C–H bonds at the α, β, or even farther positions can serve as electron donors to the metal center. This idea is supported by crystallographic evidence and NMR chemical shifts (the donating hydrogens shift to high field). Such interactions are called agostic interactions, and they resemble an “interrupted” transition state for hydride elimination. Alkyl complexes that cannot undergo β-hydride elimination for electronic reasons (high oxidation state, d0 metals) and vinyl complexes commonly exhibit this phenomenon. The fact that β-hydride elimination is slow for d0 metals—agostic interactions are seen instead—suggests that back-donation from a filled metal orbital to the σ*C–H is important for β-hydride elimination. Here’s an interesting, recent-ish review of agostic interactions.
In the next post in this series, we’ll explore the synthesis of metal alkyl complexes in more detail, particularly clarifying the question: how can we conquer β-hydride elimination?
Part 2:
In this post, we’ll explore the most common synthetic methods for the synthesis of alkyl complexes. In addition to enumerating the reactions that produce alkyl complexes, this post will also describe strategies for getting around β-hydride elimination when isolable alkyl complexes are the goal. Here we go!
Properties of Stable Alkyl Complexes
Stable alkyl complexes must be resistant to β-hydride elimination. In the last post we identified four key conditions necessary for elimination to occur:
1. The β-carbon must bear a hydrogen.
2. The M–C and C–H bonds must be able to achieve a syn coplanar orientation (pointing in the same direction in parallel planes).
3. The metal must bear 16 total electrons or fewer and possess an open coordination site.
4. The metal must be at least d2.
Stable alkyl complexes must violate at least one of these conditions. For example, titanium(IV) complexes lacking d electrons β-eliminate very slowly. The complex below likely also benefits from chelation (see below).
Complexes have been devised that are unable to achieve the syn coplanar orientation required for elimination, or that lack β-hydrogens outright. A few examples are provided below—one has to admire the cleverness of the researchers who devised these complexes. The metallacyclobutane is particularly striking!
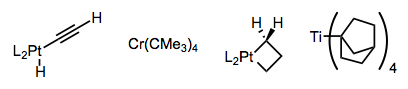
Using tightly binding, chelating ligands or a directing group on the substrate, the formation of 16-electron complexes susceptible to β-hydride elimination can be discouraged. Notice how the hyrdrogen-bonding L2 ligands in the central complex below hold the metal center in a death grip.
Finally, it’s worth noting that complexes with an open coordination site—such as 16-electron, square-planar complexes of Ni, Pd, and Pt important for cross-coupling—are susceptible to reactions with solvent or other species at the open site. Bulky alkyl ligands help prevent these side reactions. In the example below, the methyl groups of the o-tolyl ligands extend into the space above and below the square plane, discouraging the approach of solvent molecules perpendicular to the plane.
Many transition metal complexes catalyze (E)/(Z) isomerization and the isomerization of terminal alkenes (α-olefins) to internal isomers via β-hydride elimination. This is a testament to the importance of this process for alkyl complexes. Of course, transient alkyl complexes may appear to be susceptible to β-hydride elimination, but if other processes are faster, elimination will not occur. Thus, the optimization of many reactions involving alkyl complexes as intermediates has involved speeding up other processes at the expense of β-hydride elimination—hydrocyanation is a good example.
Synthesis of Alkyl Complexes
The dominant synthetic methods for alkyl complexes are based on nucleophilic attack, electrophilic attack, oxidative addition, and migratory insertion. The first two methods should be intuitive to the organic chemist; the second two are based on more esoteric (but no less important) reactions of organometallic complexes.
Metals bearing good leaving groups are analogous to organic electrophiles, and are susceptible to nucleophilic attack by organolithiums, Grignard reagents, and other polarized organometallics. These reactions can be viewed as a kind of transmetalation, as the alkyl ligand moves from one metal to another. Electron-withdrawing X-type ligands like –Cl and –Br should jump out as good leaving groups. On the other hand, clean substitution of L-type ligands by anionic nucleophiles is much more rare (anionic complexes would result).
Many anionic metal complexes are nucleophilic enough to attack electrophilic sources of carbon such as alkyl and acyl halides in an electrophilic attack mode. An available lone pair on the metal and open coordination site are prerequisites for this chemistry. The charge on the complex increases by one unit (in effect, negative charge is transferred to the electrophile’s leaving group). We can classify these as oxidative ligation reactions—notice that the oxidation state of the metal increases by two units.
Oxidative addition results in the cleavage of a W–Z bond and placement of two new X-type ligands (–W and –Z) on the metal center, with an increase in the oxidation state of the metal and the total electron count by two units. Organic halides are extremely common substrates for this reaction, the first step in the mechanism of cross-coupling reactions. The oxidized metal complex containing new alkyl and halide ligands is the final product. Notice that two open coordination sites are required (not necessarily simultaneously), the metal center must be amenable to two-electron oxidation, and the number of total electrons of the complex increases by two. In essence, the electrons of the W–Z bond join the complex’s party. Take note that there are many known mechanisms for oxidative addition! We’ll explore these different mechanisms in detail in a future post.
Finally, migratory insertion of unsaturated organic compounds is an important method for the synthesis of certain alkyl complexes, and an important step of organometallic reactions that result in addition across π bonds. Migratory insertion is the microscopic reverse of β-hydride elimination. The clever among you may notice that the use of migratory insertion to synthesize alkyl complexes seems inconsistent with our observation that its reverse is ubiquitous for metal alkyls—shouldn’t equilibrium favor the olefin hydride complex? In many cases this is the case; however, there are some notable exceptions. For example, perfluoroalkyl complexes are exceptionally stable (why?), so the insertion of fluoroalkenes is often favored over elimination.
As we noted above, we can still invoke kinetically stable alkyl complexes as intermediates in reactions provided subsequent steps are faster. In the next post, we’ll examine the general classes of reactions in which alkyl complexes find themselves the major players, focusing on the specific mechanistic steps that involve the alkyl complex (reductive elimination, transmetalation, migratory insertion, and [naturally] β-hydride elimination).
Part 3:
In this last post on alkyl ligands, we’ll explore the major modes of reactivity of metal alkyls. We’ve discussed β-hydride elimination in detail, but other fates of metal alkyls include reductive elimination, transmetallation, and migratory insertion into the M–C bond. In a similar manner to our studies of other ligands, we’d like to relate the steric and electronic properties of the metal alkyl complex to its propensity to undergo these reactions. This kind of thinking is particularly important when we’re interested in controlling the relative rates and/or extents of two different, competing reaction pathways.
Reactions of Metal Alkyl Complexes
Recall that β-hydride elimination is an extremely common—and sometimes problematic—transformation of metal alkyls. Then again, there are reactions for which β-hydride elimination is desirable, such as the Heck reaction. Structural modifications that strengthen the M–H bond relative to the M–C bond encourage β-hydride elimination; the step can also be driven by trapping of the metal hydride product with a base (the Heck reaction uses this idea).
On the flip side, stabilization of the M–C bond discourages elimination and encourages its reverse: migratory insertion of olefins into M–H. Previously we saw the example of perfluoroalkyl ligands, which possess exceptionally stable M–C bonds. The fundamental idea here—that electron-withdrawing groups on the alkyl ligand stabilize the M–C bond—is quite general. Hartwig describes an increase in the “ionic character” of the M–C bond upon the addition of electron-withdrawing groups to the alkyl ligand (thereby strengthening the M–C bond, since ionic bonds are stronger than covalent bonds). Bond energies from organic chemistry bear out this idea to an extent; for instance, see the relative BDEs of Me–Me, Me–Ph, and Me–CCH in this reference. I still find this explanation a little “hand-wavy,” but it serves our purpose, I suppose.
Metal alkyls are subject to reductive elimination, the microscopic reverse of oxidative addition. The metal loses two covalent ligands, its formal oxidation state decreases by two units, total electron count decreases by two units, and an R–X bond forms. Reductive elimination is favorable when the R–X bond in the organic product is more stable than the M–R and M–X bonds in the starting complex (a thermodynamic issue). It should be noted, however, that the kinetics of reductive elimination depend substantially on the steric bulk of the eliminating ligands. Concerted reductive elimination of R–H usually possesses a lower activation energy than R–R elimination.
Transmetalation involves the transfer of an alkyl ligand from one metal to the other. An interesting problem concerns the relative reactivity of metal alkyls toward transmetalation. Assuming similar, uncomplicated ligand sets, which of two metal centers is more likely to hold on to an alkyl ligand? Consider the two situations below.
\[\ce{MR + M’ <=> M + M’R} \nonumber\]
\[\ce{MR + M’R’ <=> MR’ + M’R} \nonumber\]
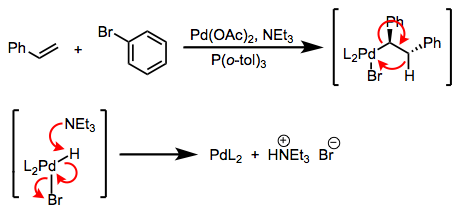
The first is a bona fide transmetalation; the second is really a double replacement reaction. The distinction is rarely drawn in practice, but it’s important! The difference is that in the first case, a single-electron transfer of sorts must take place, while in the second case, no redox chemistry is necessary. Favorability in the first case is governed by the relative reduction potentials of M and M’ (the reaction goes forward when M’ is more easily oxidized than M); in the second case, the relative electropositivities of the metals is key, and other factors like lattice energies may be important. The distinction between transmetalation per se and double replacement explains the paradoxical synthetic sequence in the figure below. In practice, both are called “transmetalation.” See these slides (page 6) for a summarizing reference.
This brings our extended look at metal alkyl complexes to a temporary close. Of course, metal alkyls are everywhere in organometallic chemistry…so seeing them again is pretty much inevitable! The next installment in the Epic Ligand Survey series concerns allyl, cyclopentadienyl, and other odd-membered pi systems. These LnX-type ligands can, like arenes, pile as many as six electrons on the metal center at once.