
Heck Reaction
- Page ID
- 69095
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)The Heck reaction is a famous chemical reaction discovered by Mizoroki and Heck in 1972 through independent research. It involves the cross-coupling reaction between organohalides and alkenes, these two substances react in the presence of a palladium catalyst and a base to form a substituted alkene:
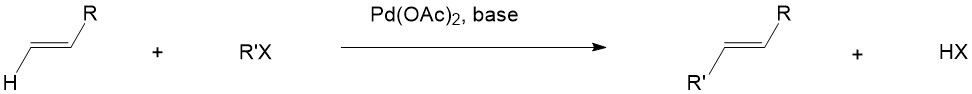
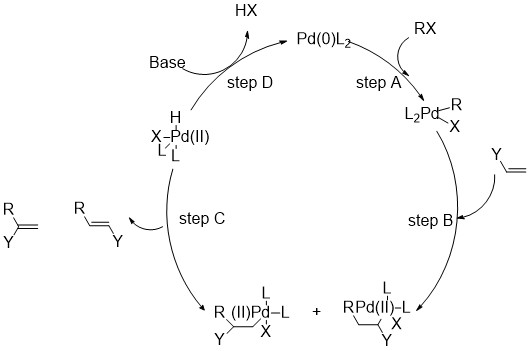
Step A is the oxidative addition of a polar substrate onto a palladium catalyst to form a tetrasubstituted complex. Step B is the migratory insertion of an olefin into the system. Step C is the β-Hydride Elimination of the alkene and Step D is the addition of base to the palladium to regenerate the starting catalyst and close the cycle[1].
Detailed Study of the Mechanism of Heck Reaction
Pre-activation of Palladium Catalyst
It is noteworthy that the first step of the catalytic cycle is actually not the oxidative addition of the substrate, as the palladium catalyst must be activated before the reaction. Therefore, a thorough study of the structure of the palladium catalyst and its properties will be important in understanding Heck reactions [2]. The catalytic precursor Pd(II)(OAc)2, associated with monodentate phosphine ligands such as PPh3 is normally used to catalyze the reaction, but this Pd(II) complex must be reduced to Pd(0) in order to enter the catalytic cycle. There are two different mechanisms involving phosphine-mediated Pd(II) reduction[2]:


The catalytic precursor Pd(II)(OAc)2, associated with monophosphine ligands, is much more efficient in catalyzing Heck reactions when compared to Pd(0)(PPh3)4 catalyst. This is because Pd(0)(PPh3)2(OAc)- can be destabilized by the interaction between OAc- and protons to readily form the unstable Pd(0)(PPh3)2 catalyst, which then enters the catalytic cycle to catalyze the reaction[3]. Since Pd(0)(PPh3)4 is a relatively stable 18-electron complex, it is unlikely that it dissociates two ligands to form an unstable 14-electron structure[3].

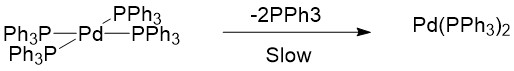
Furthermore, Pd(0) catalysts must possess an appropriate coordination number to enter the catalytic cycle[2]. If there are too many monophosphine ligands, it may inhibit the catalyst because a coordinatively saturated metal complex will be formed via ligand association:
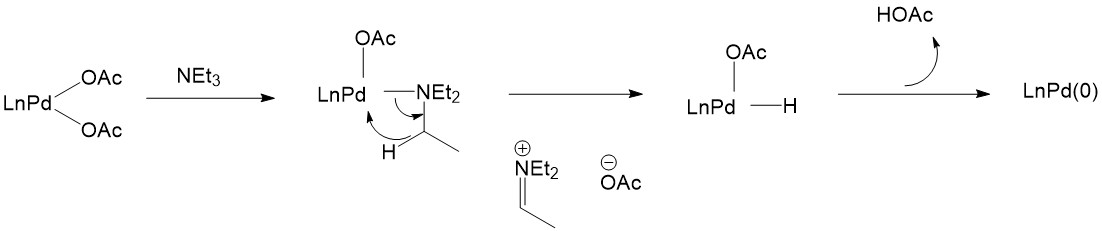
The activation of Pd(II) catalysts can also be achieved without the assistance of phosphines. For example, triethylamine is a good reagent to selectively reduce Pd(II):
Oxidative Addition
Oxidative addition is the most difficult step of the entire catalytic cycle. However, the presence of electron-donating groups on the phosphine ligands can activate the Pd(0) catalyst such that the R-X bond can be easily broken along with the formation of Pd-R and Pd-X bond. The rate of oxidative addition also depends on the chemical property of halides, the following trend is generally observed [2]:
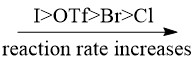
Olefin Addition to the Palladium Complex
Before migratory insertion of the olefin to the palladium-R bond, the olefin must first associate onto the palladium complex, which requires the dissociation of the existing ligands. Historically, the Heck reaction was viewed as the functionalization of olefins through aryl halides typically without ligands for aryl iodide or with monodentate phosphines for the other compounds [1]. This cycle produces a strong Pd-X bond while there is a weak Pd-PR3 attachment. The oncoming group would attack and form a net neutral square planar molecule, as is shown in Path A. This reaction mechanism was found to not be ideal as, “chelating diphosphines… in general do not produce useful catalysts [1].” This concern was addressed through the introduction of triflates as leaving groups which would allow the mechanism coordination-insertion step to follow Path B. Catalytic and stoichiometric studies have been utilized to determine these pathways.
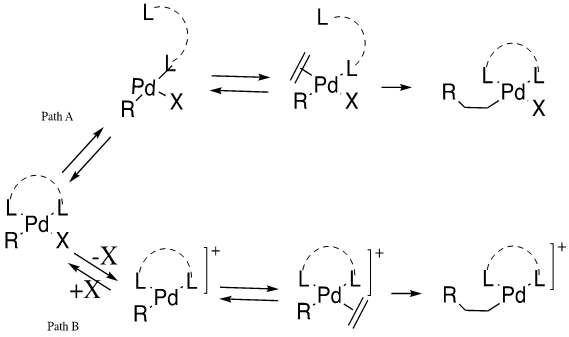
- Path A: If a neutral ligand dissociates, then the neutral mechanism predominates the reaction. The neutral mechanism occurs when X=Cl, Br, I (i.e. strong sigma-donor).
- Path B: If an anionic halide ligand dissociates, then the cationic mechanism dominates the reaction. The cationic mechanism is believed to happen when X= OTs- or OAc- (e.g. weakly associated ligand).
The two mechanisms shown above display the possible coordination-insertion paths found in Heck reactions. It has been found that the insertion of ethylene into the Pt-H bond is critical in the reaction characterization [1]. Thorn and Hoffman conducted orbital studies to determine key information from this step. The first conclusion they reached is that there must be a coplanar assembly of the metal, hydride and ethylene for insertion to occur. This indicated that insertion is stereoselective and occurs in a syn manner. Experimental data supported this claim [1]. They also determined that the energy barrier for a pentacoordinated complex is significantly higher than that of a tetracoordinated complex, indicating pentacoordinated complexes are not involved in the mechanism. This observation was supported by kinetic and experimental data [1].
Path B was unknown until 1991, when Ozawa and Hayashi proposed the existence of a cationic form of the square planar complex. To obtain this complex, triflates must be used as the leaving group [1]. The lability of Pd-OTf bonds in the oxidation addition complex aids in this formation. Bidentate phosphorus or nitrogen ligands, along with the triflate leaving group, allow for the reaction to follow this path. It was also determined that this path can yield high asymmetric induction when the diphosphine is chiral. This effect was not seen in Path A [1].
Ligand dissociation mechanisms are also affected by different types of phosphine ligands. Monodentate phosphine ligands lead to the occurrence of both the neutral and cationic mechanisms, whereas bidentate phosphine ligands merely induce a cationic mechanism, but the neutral mechanism is still possible in the presence of a large bite angle [2].
Migratory Insertion
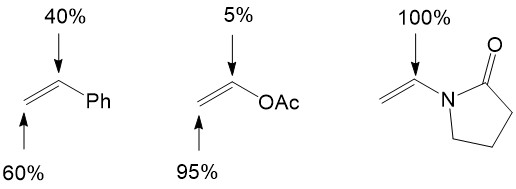
Migratory insertion of the olefin into the Pd-R bond is a crucial step for the catalytic cycle because it can control the stereo-selectivity and regio-selectivity of Heck reactions. For a neutral palladium complex, the regioselectivity is governed by sterics, which means nucleophilic attack happens on the less hindered site of the alkene [4][5]:

For cationic palladium complexes, the regioselectivity is governed by electronics, which implies that nucleophilic attack occurs on the site possessing the least electron density of the alkene [4][5]:
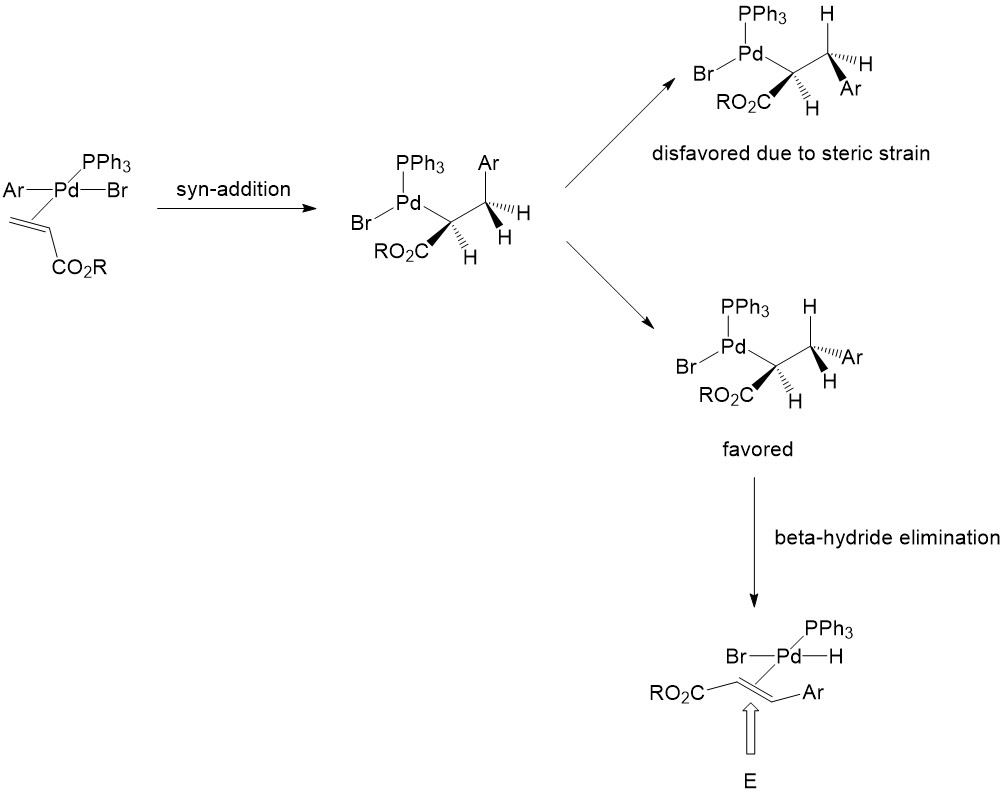
β-Hydride Elimination
β-Hydride Elimination results in the Heck reaction product, which is a new substituted alkene. In this step, the palladium and the hydride attached to it must be syn-coplanar for the initiation of elimination. The product with the Z-conformation is strongly disfavored because of the steric interaction in the transition state [2]:
Afterβ-hydride elimination, the newly formed palladium-alkeneπcomplex is subject to olefin isomerization, resulting in the formation of an undesired Heck product[4]:

These side reactions will occur since this is a reversible reaction. If the olefin dissociation rate is too slow, this problem arises [1]. Fortunately, adding bases or silver salts can significantly reduce the chance of alkene isomerization by facilitating reductive elimination to form an H-X bond [6].
Regeneration of palladium catalyst
The addition of base is necessary to reduce the L2PdHX complex back to the starting L2Pd(0)[1]. Some common bases used are trialjylamines such as Et3N or inorganic salts such as AcONa. A proton sponge or Tl(I) or Ag(I) salts may also be employed to close the cycle[1].
Intramolecular Heck Reaction
Heck reactions can also be performed in a single molecule which is quite useful for macrocyclization. This intramolecular Heck reaction was first reported by Mori and Ban in 1977:
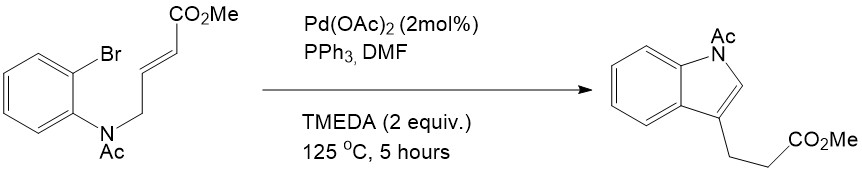
Mori, M; Ban, K.; Tetrahedron 1977, 12, 1037
The Intramolecular Heck reaction has many advantages compared with the intermolecular Heck reaction. First of all, only mono- or disubstituted alkenes can coordinate into the palladium complex in the intermolecular Heck reaction, whereas tri- and tetrasubstituted alkenes are able to participate readily through the intramolecular mechanism. Secondly, the intramolecular Heck reaction is much more efficient than the intermolecular reaction because of entropic considerations [7]. Finally, regioselectivity and stereoselectivity are dramatically improved in the intramolecular Heck reaction. This advantage inspired both Shibasaki and Overman to explore the asymmetric effect in the intramolecular Heck reaction, and they eventually found the first asymmetric intramolecular Heck reactions. This remarkable finding has provided enlightenment for natural product synthesis [7].
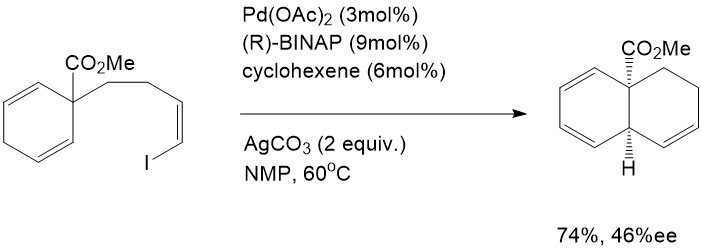
Shibasaki, M.J.Org. Chem. 1984,54,4738
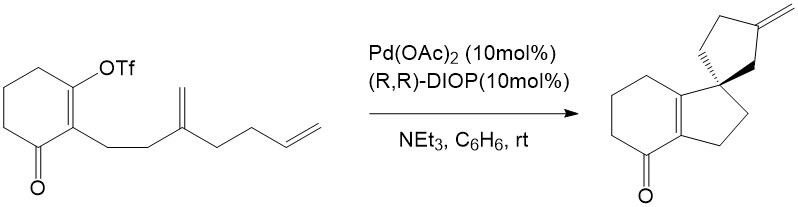
Overman, L. E. J. Org. Chem. 1989, 54, 5846
Regioselectivity and Stereoselectivity
Regioselectivity reactions of Path A were conducted on several classes of olefins. Different olefin families reacted with different regioselectivity[1]. Once aryl triflates were introduced as the leaving groups, reactions via Path B were also examined for regioselectivity. These reactions were carried out with aryl triflate leaving groups and aryl halides with Pd(OAc)2 with bidentate phosphorus ligands[1]. These reactions yielded branched products more readily than Path A.
In Path A, regioselectivity is related to the coordination-insertion pathway and steric factors. A migration of the R group to the less substituted carbon with formation of linear products has seen to be favored in Pathway A [1]. Path B differs in that electronic factors dominate in determining regioselectivity. The increase in polarization is determined by the coordination of a pi-system within a cationic complex. This causes selective migration of the aryl moiety onto the carbon with a lower charge density [1].
Hayashi and Ozawa have investigated stereoselective intermolecular reactions using aryl triflate as the leaving group and chiral (R)-BINAP. Several reactions were performed under analogous conditions and their products compared [1]. As is expected with the triflate leaving group, these reactions followed Path B. In these reactions, the chiral BINAP ligand binds tightly to the metal through both of its phosphorus atoms. This chiral ligand is able to transfer its chiral information from the catalyst to the substrates [1]. These reactions have been found to be most selective when using both chiral BINAP and the triflate leaving group. Of note is that this (R)-BINAP determination of selectivity will only occur in systems that are electron rich. This information aligns with the assertion that Path B relies heavily on electronic factors in that electron rich systems react much more efficiently than electron poor [1].
Overman has reported a stereoselective synthesis for quaternary carbons through asymmetric intramolecular Heck reactions. These results were of note, as they did not follow the widely held belief that long reaction times result in isomerization of the double bond and multiple product formation [1]. PMP was used as the base in these reactions without Ag(I) salt present. Good selectivity was observed in these reactions despite the slow reaction time and the difficulty in transferring chirality through Path A [1]. It was determined that with flexible substrates, the products were almost racemic. However, with rigid substrates, the single phosphorus coordination of (R)-BINAP was still able to transfer the chirality via Path A[1].
Limitations of the Heck Reaction
The Heck reaction is widely used in the pharmaceutical, medical and industrial areas because of its ability to efficiently generate large polycyclic structures. However, this synthetically useful reaction has its own disadvantages [8].One of the major disadvantages is that the Pd catalyst will be lost at the end of the catalytic cycle, therefore, it is necessary for researchers to find an effective method to recycle the palladium catalyst. Another significant weakness is that the phosphine ligands attached to the palladium catalyst can be toxic and expensive, so the phosphine-free ligands must be discovered to improve the efficiency of the reaction [8].
Applications of the Heck Reaction
• Heck couplings with dehydrocostus lactone and aryl halides can produce guaianolide sesquiterpene lactones derivatives, which have been proved effective in inhibiting resistant acute leukemic cells:
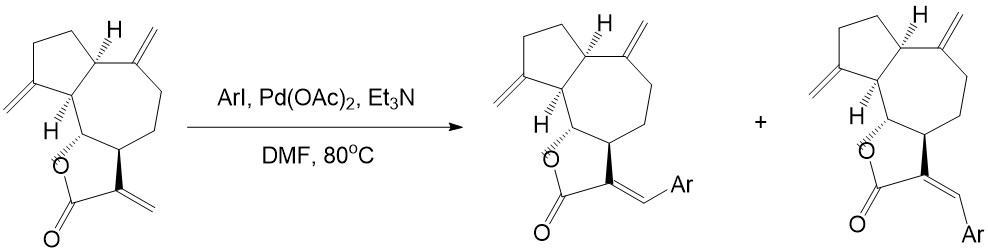
Y. H. Ding, H. X. Fan, J. Long, Q. Zhang, &Y. Chen, Bioorganic and Medicinal Chemistry Letters, vol. 23, 22, 6087–6092, 2013.
• The Heck reaction can also be used to synthesize the smoking cessation aid, Chantix ®
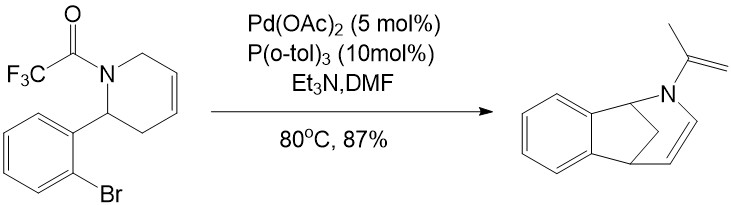
Coe, J. W.; Brooks, P.R.; Veteline, M.G.; Bashore, C.G.;Bianco, K.;Flick, A,A.C. Tetrahedron Lett. 2011, 52, 953-954
References
- Cabri, W.; Candiani, I. Acc. Chem. Res. 1995, 28, 2-7.
- Tambar, U. K. (2003, April 17). The Heck Reaction:Mechanistic Insight into a Synthetically Useful Reaction. Stoltz Group Literature Series.
- Jutand, A. (2009, February 17). Mechanism of the Mizoroki-Heck Reaction. The Mizoroki-Heck Reaction, 1-50. doi: 10.1002/9780470716076.ch1
- Haidle, A., Mousseau, J., & Liu, F. (n.d.). The Heck Reaction. Retrieved December 06, 2016, from http://faculty.chemistry.harvard.edu/files/myers/files/heck-matt1.pdf
- Cabri, W.; Candiani, I.; Bedeschi, A.; Penco, S.; Santi, R .J. Org. Chem. 1992, 57, 1481-1486
- Abelman, M. M.; Oh, T.; Overman, L. E. J. Org. Chem. 1987, 52, 4133-4135
- Knowles, R. (2004, July 14). The Intramolecular Heck Reaction, MacMillan Group Meeting
- Zazybin, A. (2010, January 22). Organometallic Chemistry and Homogeneous Catalysis. Retrieved December 06, 2016, from http://sas.k.u-tokyo.ac.jp/AZ/Lecture12.pdf
Contributors and Attributions
- Haley Merritt and Yifan Qi