
Cross-Dehydrogenative Coupling
- Page ID
- 69093
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Cross-dehydrogenative coupling (CDC) is the class of reaction developed by Chao-Jun Li (McGill U)17 that results in the formation of C-C or C-N bond directly from two unmodified C-H bonds (C-C bond formation) or C-H and N-H bonds (C-N bond formation) (Figure 1). Formally, the reaction occurs with a loss of an equivalent of H2, although hydrogen gas is really the byproduct since the formation of C-C or C-N bond with a loss of H2 is thermodynamically unfavorable and thus requires the use of an oxidant (Cu(ІІ), Ag(І) salts, CAN, benzoquinone, peroxides, O2, hypervalent iodine, persulfates etc.) as the driving force.
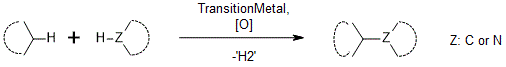
Although the same transformations can be achieved by alternative methods, the advantage of CDC approach is that direct tandem oxidation of simple C-H and N-H bonds allows use of simple and readily available reagents and reduces number of steps towards the desired molecule. On the other hand, some challenges are yet to be overcome including low reactivity of some C-H bonds, overoxidation, selective C-H bond functionalization, dimerization etc.
CDC reactions are used to construct bonds between sp3-sp3, sp3-sp2, sp3-sp, sp2-sp2 (including Heck-type reaction), sp2-sp (Sonogashira type) and sp-sp (Glaser reaction) carbon atoms, as well as C(sp3)-N. Because CDC is a group of reaction in which the mechanism and reactivity various dramatically depending on the substrate. These couplings, however, can be roughly divided in four groups (by the proposed mechanism): CDC via Heck-type mechanism, direct arylation, CDC via ionic intermediates and CDC via radical intermediates.1
CDC via Heck-type Mechanism
Heck-type mechanism2 is typical of sp2-sp2 CDCs including arene-alkene, alkene-alkene, arene-benzoquinone coupling; sp3-sp2; and formal sp3-sp2 including enolate-alkene, allyl-arene coupling. As follows from the name, the mechanism is very similar with the Heck Reaction. Palladium3 salts are usually the catalyst of choice, although the same reactivity may be achieved with other transition metals such as Rh(ІІ), Ru(III) and Ir(І) complexes. The mechanism involves the electrophilic palladation of arene ring (ligand assisted or base mediated C-H insertion) with a Pd(II) catalyst to generate arylpalladium intermediate. Subsequent carbopalladation of olefin leads to alkylpalladium complex that undergoes syn-β-H elimination to yield styrenyl product and Pd(0). The final step of the catalytic cycle is oxidation of Pd(0) to Pd(ІІ). In an alternative mechanism, the Pd(ІІ) catalyst is proposed to coordinate to the olefin, which enhances its electrophilicity and propensity to undergo nucleophilic addition with electron-rich aromatic rings (Figure 2).
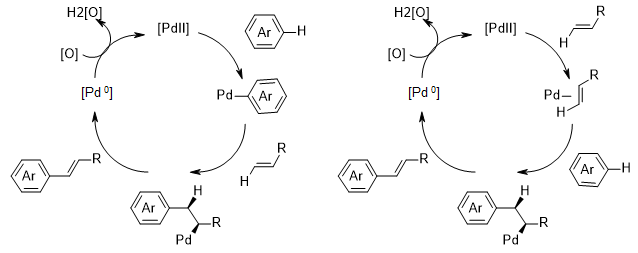
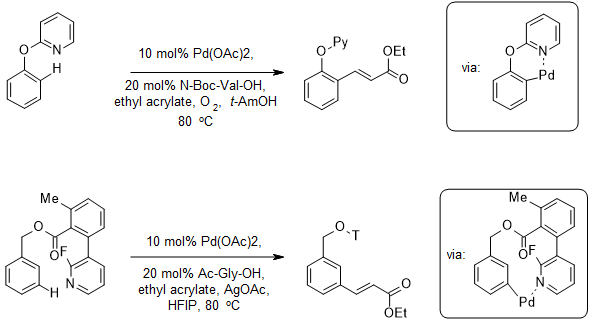
Catalytic systems are usually not too complex and require no or only a simple ligand such as phosphines, N-protected amino acids or sulfoxides. Inorganic or organic base can also be employed. When more than one product may be formed, the regeoselectivity is normally achieved by installation of a directing group on the aromatic ring, whereas the pyridyl-based groups are the most extensively employed to direct transition-metal-catalyzed C−H activation reactions (both ortho- and meta-).4
Diract Arylation
Direct arylation (also known as 2-fold C-H activation) is used to construct a bond between two arenes.5 Direct arylation strategy is an alternative to such well-known and widely used methods as: Suzuki, Negishi, Stille, Hiyama, Kumada cross-couplings etc, which require independent synthesis and isolation of aryl halide (or pseudohalide) and arylmetal starting materials. Direct arylation can be achieved with electron-rich arenes, arenes bearing pyridine, amide, carbamate, and other oxazoline directing groups, pyridine N-oxides and other electron-deficient arenes. Palladium(ІІ) complexes are almost exclusively used. The method is considered as atom economical approach towards bi- and poly-aryl motifs, which are common structures in medicinal chemistry and material science. The mechanism involves 2-fold C-H activation (either ligand assisted or base mediated) leading to the formation of Pd(ІІ) bis-aryl complex, which undergoes reductive elimination and produces biaryl and Pd(0). The final step of the catalytic cycle is oxidation of Pd(0) to Pd(ІІ) (Figure 4).

The regeoselectivity is usually controlled by thermodynamic factors (e.g. formation of the most stable intermediate through activation of the most acidic C-H bond or through the attack of the most nucleophilic position, Figure 5a)6 and by installation of a directing group (Figure 5b).7
CDC via Ionic Intermediates
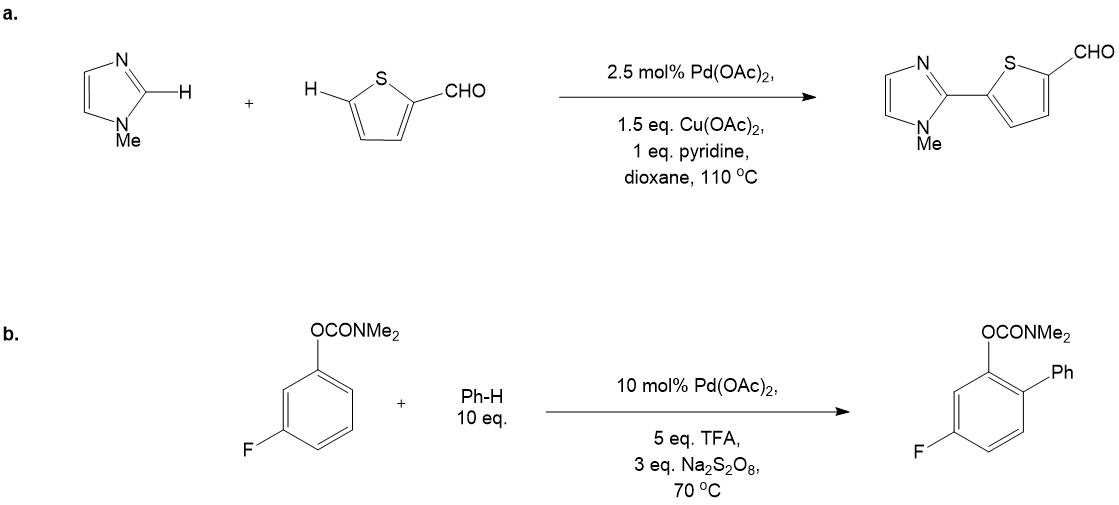
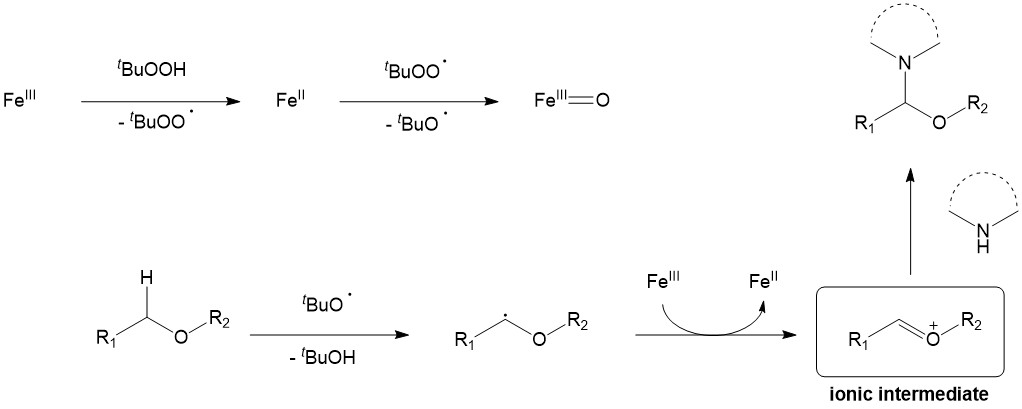
Unlike first two mechanisms CDC via ionic intermediates can be promoted by a number of transition metals including Pd, Ru, Zr, Ni, Cu, Fe, Co etc., whereas the last three are the most widely employed. The ionic mechanism predominantly relies on single electron transfer (SET) pathway, although some exceptions are known.8 Mechanistically C-C/N bond formation occurs between an electrophilic carbon species (carbocation) and a carbon- or nitrogen-based nucleophile (amine, amide, carbanion, enamine, heteroaromatic). Carbocations are generated in situ by C-H bond oxidation of a suitable precursor, whereas carbanions can be prepared by deprotonation of relatively acidic C-H or N-H bonds. As a source of carbocations, amines, ethers, allylic and benzylic compounds are usually employed. Oxidant choice depends on a substrate, although organic hydroperoxides were shown to be most efficient in many cases. The catalysis normally performed without ligand additive and simple TM salts are employed (halides, carboxylates, nitrates etc.) While it is hard to derive a single mechanism, the Ionic Intermediates pathway can be demonstrated on the iron(III) catalyzed CDC of ethers with N-nucleophiles, leading to a facile formation of a hemiaminal moiety (which present in a number of bioactive natural products) (Figure 6).
CDC via Radical Intermediates
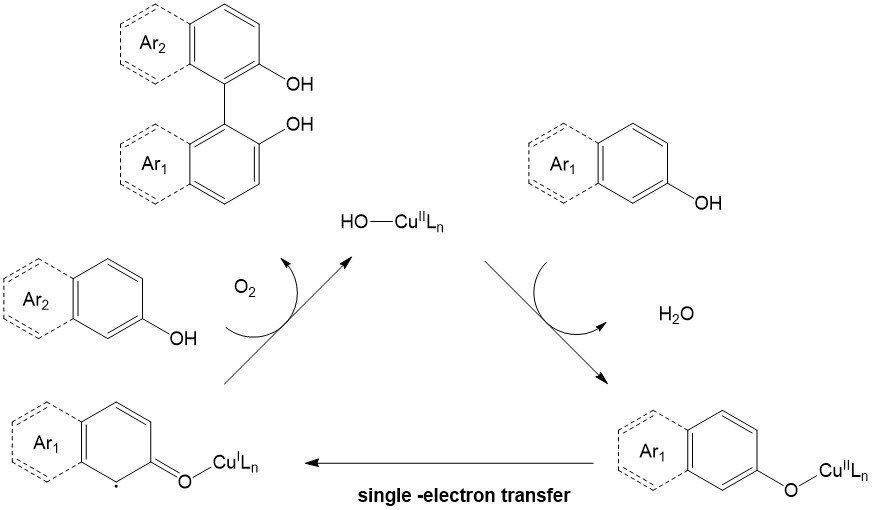
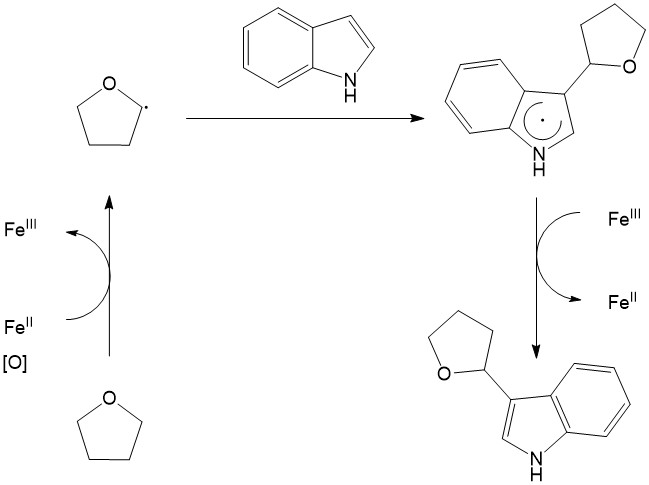
The radical Intermediates mechanism is very similar to the one described above (Ionic), except that no ionic intermediates are formed. The newly generated carbon-centered radical directly reacts with another active specie (e.g. carbocation, carboanion, C-C multiple bond) forming another radical, which later on gets oxidized either by an oxidant (Figure 7)9 or transition metal (Figure 8)10 realizing the desired product. The radical CDC is exclusively promoted by first-row transition metal complexes such as Cu, Fe, Mn (in some cases vanadium oxo-species) via single-electron transfer type pathway. Selective C-H bond oxidation to a radical can be achieved with phenols, electron-rich arenes, benzylic compounds and C(sp3)-H bond α- to carbonyl or heteroatom (O or N).
The radical CDC is essential in the synthesis of binaphtols11 and related compounds and can be achieved enantioselectively when chiral ligand is used (i.e. Salan, (R)-α-methylbenzylamine etc.).12
Cross-dehydrogenative coupling is a rapidly advancing field and may occur via a wide array of pathways. Therefore some valuable transformations including Au-catalyzed dehydrogenative cyclization between allenes and arenes,13 Cu and Ni-catalyzed cross-coupling of alkynes,14 Cu and Pd-catalyzed CDC of aldehydes and arenes making ketones etc.,15 were not described. In many cases involving C(sp3)-H and sometimes C(sp2)-H bonds CDC may occur without transition metal catalyst. Finally, it is worth noting, that CDC approach is widely used by nature to construct C-C bonds in living organisms, which primarily involves catalysis promoted by cytochrome P450 (CYP) enzymes in the presence of O2 as the terminal oxidant.16
References
- Yeung, C. S.; Dong, V. M., Catalytic Dehydrogenative Cross-Coupling: Forming Carbon-Carbon Bonds by Oxidizing Two Carbon-Hydrogen Bonds. Chem Rev 2011, 111 (3), 1215-1292.
- Beccalli, E. M.; Broggini, G.; Martinelli, M.; Sottocornola, S., C-C, C-O, C-N bond formation on sp(2) carbon by Pd(II)-catalyzed reactions involving oxidant agents. Chem Rev 2007, 107 (11), 5318-5365.
- Huang, H.; Stewart, T.; Gutmann, M.; Ohhara, T.; Niimura, N.; Li, Y. X.; Wen, J. F.; Bau, R.; Wong, H. N. C., To Flip or Not To Flip? Assessing the Inversion Barrier of the Tetraphenylene Framework with Enantiopure 2,15-Dideuteriotetraphenylene and 2,7-Dimethyltetraphenylene. J Org Chem 2009, 74 (1), 359-369.
- Ling Chu, M. S., Keita Tanaka, Qinghao Chen, Natalya Pissarnitski, Eric Streckfuss, Jin-Quan Yu, Remote Meta-C−H Activation Using a Pyridine-Based Template: Achieving Site-Selectivity via the Recognition of Distance and Geometry. ACS Cent. Sci. 2015, (1), 394−399.
- (a) Lyons, T. W.; Sanford, M. S., Palladium-Catalyzed Ligand-Directed C-H Functionalization Reactions. Chem Rev 2010, 110 (2), 1147-1169; (b) Ashenhurst, J. A., Intermolecular oxidative cross-coupling of arenes. Chem Soc Rev 2010, 39 (2), 540-548.
- Xi, P. H.; Yang, F.; Qin, S.; Zhao, D. B.; Lan, J. B.; Gao, G.; Hu, C. W.; You, J. S., Palladium(II)-Catalyzed Oxidative C-H/C-H Cross-Coupling of Heteroarenes. J Am Chem Soc 2010, 132 (6), 1822-+.
- Zhao, X. D.; Yeung, C. S.; Dong, V. M., Palladium-Catalyzed Ortho-Arylation of O-Phenylcarbamates with Simple Arenes and Sodium Persulfate. J Am Chem Soc 2010, 132 (16), 5837-5844.
- (a) Li, Z. P.; Li, C. J., Catalytic allylic alkylation via the cross-dehydrogenative-coupling reaction between allylic sp(3) C-H and methylenic sp(3) C-H bonds. J Am Chem Soc 2006, 128 (1), 56-57; (b) Lin, S.; Song, C. X.; Cai, G. X.; Wang, W. H.; Shi, Z. J., Intra/intermolecular direct allylic alkylation via Pd(II)-catalyzed allylic C-H activation. J Am Chem Soc 2008, 130 (39), 12901-+.
- Li, X. L.; Hewgley, J. B.; Mulrooney, C. A.; Yang, J. M.; Kozlowski, M. C., Enantioselective oxidative biaryl coupling reactions catalyzed by 1,5-diazadecalin metal complexes: Efficient formation of chiral functionalized BINOL derivatives. J Org Chem 2003, 68 (14), 5500-5511.
- Guo, X. W.; Pan, S. G.; Liu, J. H.; Li, Z. P., One-Pot Synthesis of Symmetric and Unsymmetric 1,1-Bis-indolylmethanes via Tandem Iron-Catalyzed C-H Bond Oxidation and C-O Bond Cleavage. J Org Chem 2009, 74 (22), 8848-8851.
- Chen, Y.; Yekta, S.; Yudin, A. K., Modified BINOL ligands in asymmetric catalysis. Chem Rev 2003, 103 (8), 3155-3211.
- (a) Yamamoto, K.; Yumioka, H.; Okamoto, Y.; Chikamatsu, H., Synthesis and Chiral Recognition of an Optically-Active Bis-Crown Ether Incorporating a Diphenanthrylnaphthalene Moiety as the Chiral Center. J Chem Soc Chem Comm 1987, (3), 168-169; (b) Egami, H.; Matsumoto, K.; Oguma, T.; Kunisu, T.; Katsuki, T., Enantioenriched Synthesis of C-1-Symmetric BINOLs: Iron-Catalyzed Cross-Coupling of 2-Naphthols and Some Mechanistic Insight. J Am Chem Soc 2010, 132 (39), 13633-13635.
- Hopkinson, M. N.; Tessier, A.; Salisbury, A.; Giuffredi, G. T.; Combettes, L. E.; Gee, A. D.; Gouverneur, V., Gold-Catalyzed Intramolecular Oxidative Cross-Coupling of Nonactivated Arenes. Chem-Eur J 2010, 16 (16), 4739-4743.
- Su, L. B.; Dong, J. Y.; Liu, L.; Sun, M. L.; Qiu, R. H.; Zhou, Y. B.; Yin, S. F., Copper Catalysis for Selective Heterocoupling of Terminal Alkynes. J Am Chem Soc 2016, 138 (38), 12348-12351.
- Jia, X. F.; Zhang, S. H.; Wang, W. H.; Luo, F.; Cheng, J., Palladium-Catalyzed Acylation of sp(2) C-H bond: Direct Access to Ketones from Aldehydes. Org Lett 2009, 11 (14), 3120-3123.
- Denisov, I. G.; Makris, T. M.; Sligar, S. G.; Schlichting, I., Structure and chemistry of cytochrome P450. Chem Rev 2005, 105 (6), 2253-2277.
- Li, C.-J. Cross-dehydrogenative coupling (CDC): Exploring C− C bond formations beyond functional group transformations” Acc. Chem. Res. 2009, 42, 335-344.
Contributors
- Kirill Korvinson (City University of New York)