
Hydroamination
- Page ID
- 69096
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Hydroamination is a reaction that involves the addition of a hydrogen and an amino group across an unsaturated C-C bond, such as that in alkenes or in alkynes. It is a form of hydrofunctionalization with the functional group being an amine or amino derivative.
Hydroamination is most often performed catalytically. The catalysts for hydroamination are broad and varied, including acids, bases, and transition metals in both homogenous and heterogeneous media. This report will focus primarily on the homogeneous transition metal catalysis of hydroamination.
The general reaction for hydroamination is illustrated in Scheme 1.
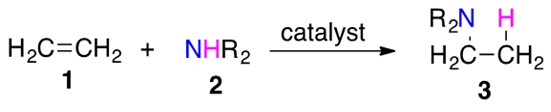
Scheme 1: Amine 2 is added across unsaturated C-C bond present in 1 to afford hydroamination product 3.
Regioselectivity for addition across asymmetrically substituted unsaturated C-C bonds is an important consideration in this reaction. Recall from general organic chemistry that regioselectivity is characterized by Markovnikov addition (to form branched products) or anti-Markovnikov addition (to form linear products). Scheme 2 builds upon the example in Scheme 1 to demonstrate the differentiating principle for these two classes of regioselective addition.
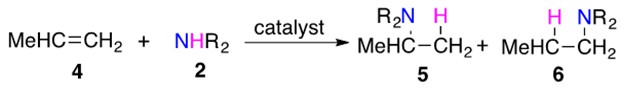
Scheme 2: Amine 2 can add across unsymmetrically substituted alkene 4 in two different ways to yield products 5 or 6.
5 is the Markovnikov product, in which the H adds to the carbon bound to the larger number of hydrogen atoms. 6 is the anti-Markovnikov product, in which H adds to the carbon bearing the least number of hydrogen atoms.
Note that hydroamination can proceed intermolecularly or intramolecularly.
This report will be divided into four different sections to discuss hydroamination based on catalyst type:
1. Lanthanide Metal Catalysts
2. Early Transition Metal (Groups 4/5) Catalysts
3. Late Transition Metal Catalysts
4. Other Catalytic Methods
Each of the three sections discussing transition metal chemistry will provide a thorough mechanistic discussion of reported hydroamination reactions, as well as illustrative examples and synthetic applications of the catalysts to total synthesis.
LANTHANIDE METAL CATALYSTS
Lanthanide or rare-earth metal catalysts for hydroamination have been studied extensively. This section will be broken down into the substrate scope of these lanthanide-metal catalysts, looking at hydroamination of aminoalkenes, aminoalkynes, aminoallenes, aminodienes. These discussions will be followed by notes on catalyst design and applications of these catalysts to total synthesis.
Aminoalkenes
We will first consider the lanthanide catalyzed hydroamination of a terminal aminoalkene (Scheme 3).
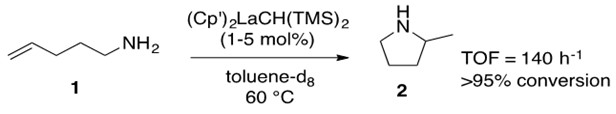
Scheme 3: Terminal amino alkene 1 is converted into cyclic amine 2 with conversion greater than 95% at a turnover frequency (TOF) of 140 h-1 through the use of lanthanide catalyst. [1]
The lanthanide catalyst used is a lanthanocene catalyst, illustrated in Figure 1.
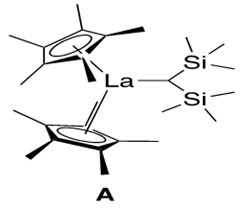
Lanthanocene catalysts such as A contain a key structural element: the metal is ‘sandwiched’ between two cyclopentadienyl rings, in a manner similar to the famous organometallic compound ferrocene. Hence the ‘-cene’ suffix is added to the metal to describe this general group of organometallic catalysts. Such nomenclature will be used throughout this report.
The reaction in Scheme 3 was found to proceed for numerous terminal alkenes containing methyl substituents on the alkyl chain for lanthanum and neodymium catalysts. The general mechanism in Scheme 4 was proposed based on kinetic experiments and isotopic labeling experiments, which are described below.
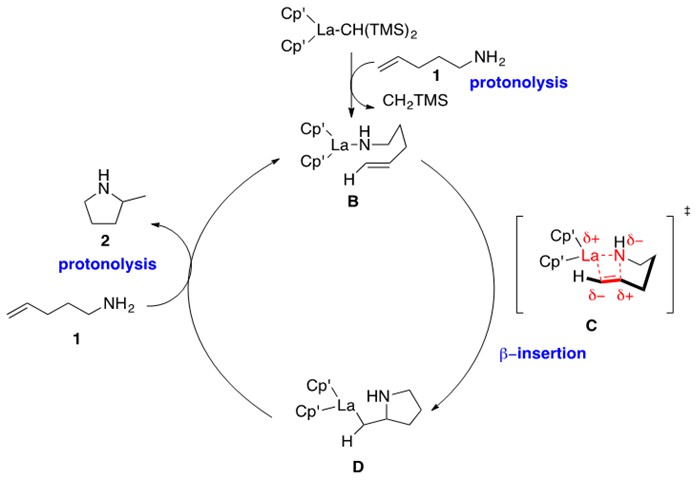
Scheme 4: Mechanism for intramolecular hydroamination (Scheme 3) catalyzed by lanthanocene catalyst A.1
This mechanism begins by protonolysis of the lanthanocene precatalyst by one equivalent of the aminoalkene to yield La-amido intermediate B. This intermediate has been characterized by single crystal X-ray diffraction data and spectroscopic data. Intermediate B undergoes b-insertion through proposed transition state C to form the La-alkyl aza-cyclic intermediate D. Note the cis relationship between the La and N atom with respect to the C-C bond, which is a key feature of these lanthanide catalyst mechanisms. This intermediate then undergoes protonolysis with a second equivalent of aminoalkene to eliminate the product cyclic amine and re-generate intermediate B. The electronic structure of the metal is consistently at the (III) oxidation state.
Kinetic studies provided a rate law that was zero-order in amine and first-order in catalyst. As suggested in Scheme 4, this indicates that the b-insertion step is the rate-determining step of the reaction. It is expected to proceed through the four-membered cyclic transition state C denoted in Scheme 4. An isotopic-labeling experiment additionally indicated that cyclization to intermediate D is irreversible.1
Catalyst design was found to influence reaction rate. As the ionic radius of the lanthanide metal increased, the rate of the reaction increased (e.g., La > Sm > Lu).1 Additionally, the less-sterically encumbered the catalyst ligation sphere, the greater the rate of the reaction (Figure 2).
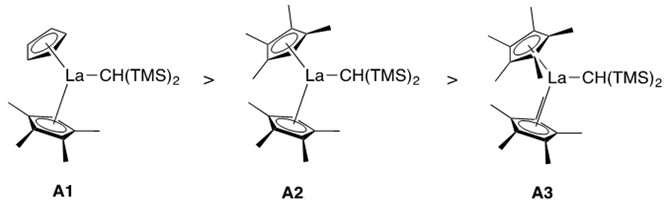
It was found that A1 reacted the fastest, with a bare cyclopentadienyl substituent and a tetramethyl-cyclopentadienyl substituent composing its lanthanocene structure. A2, where both cyclopentadienyl units are methylated at all five carbons, creating greater steric hindrance of the ligation sphere, reacted the slowest.1
One limitation of the above reported lanthanocene catalyst is that it cannot catalyze intramolecular hydroamination across substituted alkenes. This is due to unfavorable, non-bonded steric repulsions between catalyst ligands and the R groups of the alkene in the four-membered transition state C in Scheme 4.
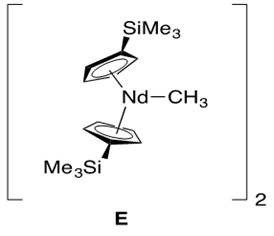
A modified lanthanide catalyst E in Figure 3 was prepared to conduct hydroamination across more hindered alkenes in an effort to reduce these non-bonded steric repulsions (Scheme 5).

Scheme 5: Reported intramolecular hydroamination across hindered alkene 3 with modified catalyst E to afford product 4. Internal alkene 5 interestingly yields no reaction.1
The key feature of catalyst E is a more open ligation environment, meaning more free space around the metal center due to less sterically bulky ligands (i.e., modified cyclopentadienyl ligands). In addition to the less sterically hindered ligation environment, these reactions required high temperature and long reaction time (many days).
Using a metal with larger ionic radius at elevated reaction temperature successfully afforded intramolecular hydroamination of internal alkenes as shown in Scheme 6. These reactions were carried out at modest TOF with excellent conversion.

Scheme 6: Intramolecular hydroamination of internal alkene 6 catalyzed by lanthanocene catalyst to afford product 7.1
Note that the reactions in Schemes 5 and 6 are all purported to follow the mechanism presented in Scheme 4.
Hence alkenes undergo intramolecular hydroamination by lanthanide-metal catalysis. Terminal alkenes are the easiest substrates with which to conduct this reaction. With internal and substituted alkenes, unfavorable steric repulsions in the transition state arise and require modified catalysts with less sterically bulky ligation environments.1
While many organolanthanide complexes facilitate intramolecular hydroamination, intermolecular lanthanide-catalyzed transformations are also possible. With the use of a phenylene-bridged binuclear organolanthanide complex F, intermolecular hydroamination has been reported (Figure 4).[2]
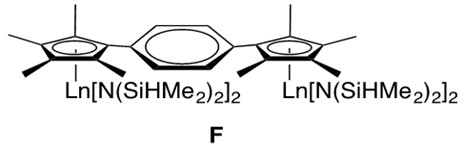
An example intermolecular hydroamination reaction with F is provided in Scheme 7.
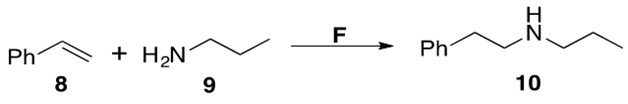
Scheme 7: Intermolecular hydroamination between styrene 8 and amine 9 to afford anti-Markovnikov product 10 using catalyst F.2
The substrate scope of F is rather limited, catalyzing reactions with only phenyl-substituted olefins or alkynes. The TOF of these reactions were low, but the yields averaged >95%. The regioselectivity of this reaction is attributed to electronic effects in the intermediate C in Scheme 4. The electronic rich phenyl substituent coordinates to the electrophilic metal center, thus forcing nucleophilic attack of the amine to take place at the terminal end of the alkenyl moiety.
Aminoalkynes
Aminoalkynes are proposed to undergo hydroamination along the same catalytic cycle as aminoalkenes (Scheme 4) because they follow the same rate law (zeroth order in amine, first order in catalyst).1
Alkynes undergo hydroamination much more readily than alkenes, which is owed to important differences in the thermodynamics of the b-insertion step: alkene insertion into the M-N bond is approximately thermoneutral whereas alkyne insertion into the M-N bond is highly exothermic.1 Given this information, we anticipate that alkyne hydroamination will avoid the limitations encountered in alkene hydroamination that had required catalyst optimization as discussed in the previous subsection. Our supposition is true: both terminal and internal alkynes readily undergo hydroamination by the same lanthanide-metal catalyst (Scheme 8). Additionally, alkyne hydroamination is accomplished much more easily and effectively than hydroamination of alkenes.
Alkyne hydroamination yields enamines for secondary amines and imines for primary amines. Examples of each reaction are reported in Scheme 8 below.
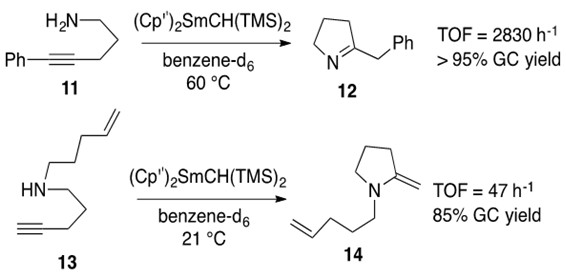
Scheme 8: Intramolecular hydroamination of internal alkyne with primary amine 11 yielding imine 12 and terminal alkyne with secondary amine 13 yielding enamine 14 using the same samarium catalyst.1
The rate-determining step in the catalytic cycle for alkyne hydroamination with lanthanide catalysts is b-insertion, as it was for alkenes.[3] In general, alkynes are found to produce higher TOFs than alkenes, which can be explained by easier b-insertion of alkynes into the M-N bond to afford the cyclic transition state H in Scheme 9 (akin to C in Scheme 4).
In lanthanide-metal catalyst design, the rate of alkyne hydroamination interestingly decreases as ionic radius of the metal increases – in stark contrast to the trend observed with alkenes.2 Thus Samarium catalysts are commonly reported metals for these transformations, as seen in Scheme 8.
Lanthanide-metal catalyzed hydroamination of alkynes has been shown to facilitate the construction of very complex azacyclic systems via tandem bicyclization, shown in Scheme 9.
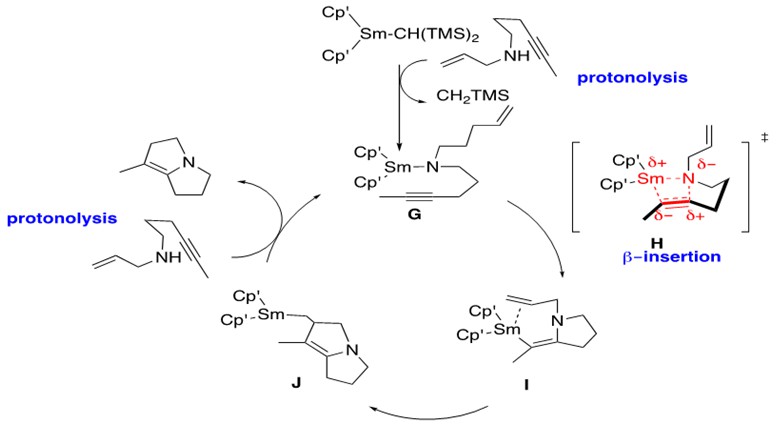
Scheme 9: Tandem bicyclization of C-N followed by C-C bond formation through intramolecular lanthanide-metal catalyzed hydroamination of an internal alkyne.1
As stated previously, the steps for hydroamination in Schemes 9 and 4 are identical. One important difference of note, however, is formation of a Sm-vinyl aza-cyclic intermediate I, in which a pi bond remains after insertion into the alkyne.
G is the catalytically active form of the Sm catalyst which undergoes rate-determining b-insertion as expected to afford I through transition state H. The additional cyclization results from insertion of the pendent terminal alkene into the Sm-C bond in I to afford J. Upon protonolysis of J with another substrate of aminoalkyne, the aza-bicyclic product is released and G is regenerated.
Aminoallenes
Allenes exhibit the same propensity for hydroamination with lanthanide metal catalysts as that of alkenes and of alkynes. Allenes follow the same general catalytic cycle for hydroamination presented in Scheme 4. Density functional theory (DFT) studies indicate that the rate-determining step for allene hydroamination is protonolysis of the Ln-vinyl aza-cyclic intermediate (akin to I in Scheme 9) rather than b-insertion to form said aza-cyclic intermediate (as is the case for alkenes and alkynes).3
Allenes notably react faster than alkenes but slower than alkynes. In terms of catalyst design, the best TOF results from lanthanide-metals of intermediate ionic radius, (Y > Sm > Lu > La).1
Aminoallene hydroamination often occurs stereoselectively which makes this transformation useful for natural product synthesis. An incredibly efficient and elegant example of such stereoselective reactions is presented in Scheme 10.
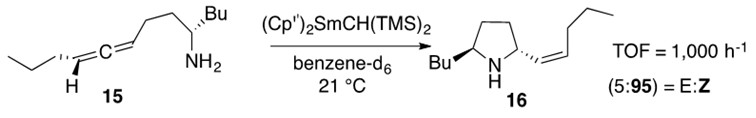
Scheme 10: Aminoallene 15 undergoes intramolecular hydroamination to afford the Z-stereoisomeric cyclic product 16 regioselectively.1
Aminodienes
A final substrate to consider is the aminodiene, whose reactivity incorporates features of the aminoallene system and the aminoalkene system. The mechanism for diene hydroamination follows that of Scheme 4, but like aminoallenes, the rate-determining step is believed to be protonolysis of the Ln-allyl aza-cyclic intermediate.3
The rate of hydroamination of aminodienes increases as the ligation environment of the catalyst becomes more open/less sterically encumbered. This indicates the same sensitivity to catalyst design as that demonstrated by aminoalkenes. Aminodienes respond to such changes in ligation environment more readily than aminoalkenes. This is best explained by the ability for the diene to datively coordinate to the metal center and form an h3-allyl intermediate L, shown and discussed in Figure 5 below.
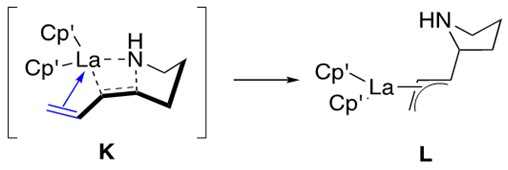
Although this allyl intermediate enhances the reaction rate, it requires a less sterically-encumbered ligation environment around the metal center in order for it to form in the first place. Thus the sensitivity of the aminodiene system to catalyst design is not owed to sterics as it was for the aminoalkene system, but rather to the ability to form the allyl intermediate.1,3
An example reaction of aminodiene hydroamination is shown below in Scheme 11.
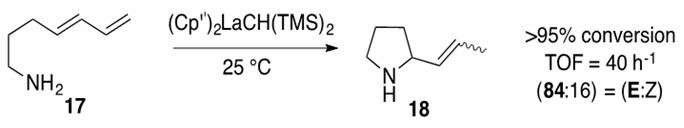
Scheme 11: Intramolecular hydroamination of aminodiene 17 with lanthanocene catalyst to afford aza-cyclic product 18.1
Additional Notes on Catalyst Design
The substrate scope of lanthanide-metal catalyzed hydroamination reactions is diverse and affords both intramolecular and intermolecular systems. These reactions follow the same general mechanism illustrated in Scheme 4, with slight variations in rate owed to differences in the ability of substrates to b-insert into the Ln-N bond and purported variations in the rate-determining step.
It should be noted that lanthanide metals generally maintain (III) oxidation state during the catalytic cycle. Formation of the catalytically active species involves replacement of a s-bonded ligand with an amine.
Sterically hindered unsaturated C-C bonds react best with more coordinatively open catalysts. This inspired the formation of half-lanthanocene catalysts, such as the samarium catalyst depicted in Figure 6, which provides solutions to the problem of ligated encumbrance retarding the catalytic reaction. This was a very important discovery in hydroamination chemistry and must be highlighted.
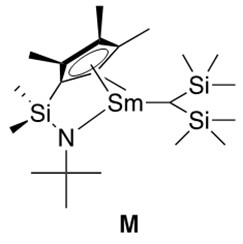
As a final note, hydroamination reactions catalyzed by actinide metals have also been reported.3
Application to Total Synthesis
Lanthanide metals are found in a variety of natural product total synthesis schemes. Consider the example in Scheme 12.
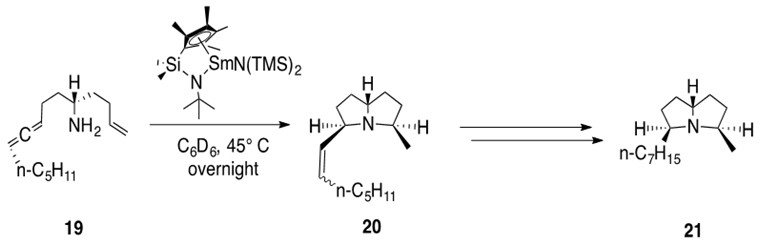
Scheme 12: Two intramolecular hydroamination reactions are catalyzed by a ‘half-lanthanocene’ samarium catalyst over an allene and an alkene moiety in 19 to afford bicyclic product 20 with cis stereochemistry. Hydrogenation of the alkene affords natural product (+)-xenovenine 21.1
Note the creation of a stereospecific, fused N-bicyclic system in one step using the remarkable catalytic hydroamination chemistry of lanthanide metals.
EARLY TRANSITION METAL (GROUPS 4/5) CATALYSTS
With Early Transition Metal Catalysts, mechanistic differences arise depending on the metal and the substrate undergoing hydroamination. Titanium and zirconium (group 4), and to a lesser extent vanadium and tantalum (group 5), have demonstrated catalytic activity for hydroamination.3
Like section 1, this section will again be divided into an analysis of the substrates undergoing hydroamination, considering alkynes, allenes, and alkenes and concluding with applications to total synthesis.
Alkynes
Both zirconium and titanium have demonstrated catalytic ability for the hydroamination of alkynes. Each metal is believed to follow approximately the same mechanistic pathway, although the rate-determining step in each catalytic cycle is interestingly proposed to be different.
Zirconium has been reported to catalyze intermolecular hydroamination between internal alkynes and primary arylamines through a bisamide complex to afford both enamine and imine product, depending on the alkyne substituents (Scheme 13).
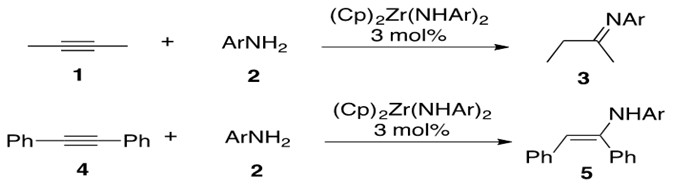
Scheme 13: Zirconium catalyzed intermolecular hydroamination of internal alkynes 1 and 4 with primary arylamine 2 affording imine 3 and enamine 5, respectively.[4]
Note that 1 initially forms a transient enamine which then rapidly tautomerizes to 3.
The following catalytic cycle (Scheme 14) was proposed for this reaction based on the data which will be presented below.

Scheme 14: Proposed catalytic cycle for zirconium bisamide catalyzed hydroamination of an internal alkyne.4
The precatalyst bisamide undergoes reversible a-elimination to generate the catalytically active Zr imido (=N) species A. Upon coordination of added alkyne 4, A undergoes [2+2] cycloaddition to form the azametallocyclobutene B. This metallocycle is then protonated upon addition of amine 2 to form C which then undergoes a-elimination to release enamine product 5 and to regenerate Zr imido species A. Note that the metal center maintains a (IV) oxidation state throughout the catalytic cycle.
Kinetic studies indicated that the reaction rate is first-order in catalyst, zeroth order in alkyne, and inverse order in amine, which is consistent with formation of Zr imido species A as the rate-determining step. One final note on this reaction is that, for asymmetrically substituted alkynes, it is proposed that [2+2] cycloaddition occurs such that the cyclic species in B contains the larger alkyne substituent a to the Zr metal. This results in an ultimately anti-Markovnikov addition of the amine substrate across the alkyne which is a commonly reported feature of this catalyst system.
Analogous to and slightly more reactive than zirconium catalysts for alkyne hydroamination are the titanium catalysts.3 Various titanocene catalysts have been studied to reach the general conclusion that the regioselectivity is determined by the bulkiness of the incoming amine and not the bulkiness of the titanium ligands. Thus a good, broadly applicable catalyst for this reaction is the bis(indole) titanocene catalyst, D, shown in Figure 7.
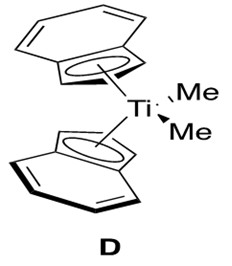
Titanocene catalysts are expected to undergo the same catalytic cycle as that depicted for zirconocene catalysts. The difference arises in the kinetic data obtained for the titanocene system, which indicates that the rate is first order in alkyne, first order in catalyst, and inverse first order in amine.3 This is different from the zirconocene data, which indicated zeroth order rate dependence in alkyne.4 This then suggests a different rate-determining step for the titanocene mechanism, which is proposed to be [2+2] cycloaddition to form the azametallocyclobutene B.
This catalyst was found to tolerate primary aryl, tert-alkyl, sec-alkyl, and linear-alkyl amines as well as internal and terminal alkynes. Terminal arylalkynes selectively formed anti-Markovnikov product while terminal alkylalkynes selectively formed Markovnikov product. This regioselectivity increased with the steric bulk of the amine, but such amines needed to be added slowly to the reaction mixture (given the inverse order rate law in amines) for sufficient reaction. Computational studies indicated that the regioselectivity of these titanocene catalysts depended on the stability of the Ti imido alkyne species prior to cyclization to afford the azametallocyclobutene B, which is largely a steric consideration.4
Advancements in catalyst design include the development of bis(amidate) titanium complexes, which are able to perform intramolecular hydroamination of aminoalkynes at room temperature. It was additionally found that mono(cyclopentadienyl) titanium (half-titanocene) catalysts catalyzed alkyne hydroamination much faster than the bis(cyclopentadienyl) titanium (titanocene) catalysts. Catalysts were designed featuring this structural trait, as shown in Figure 8.
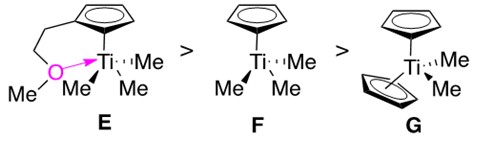
Half-titanocenes E and F react much more quickly than titanocene G. E features a pendant ether which donates into the metal, protecting it from unwanted side reactions/ coordination. The pendant ether was found to prevent undesirable dimerization of the Ti imido species A into a four-membered aggregate, which is believed to account for the significantly increased reaction rate.
Half-titanocenes are much more reactive than titanocenes, and they are considered in some reactions to be the catalytically active species (forming a Ti imido amido intermediate) rather than the bis(substituted) species.3
One key feature of this reaction is formation of the azametallocycle intermediate B. With terminal alkynes, selectivity for Markovnikov and for anti-Markovnikov hydroamination can be rationalized via steric interactions present in the formation of this intermediate. Sterically demanding amine substrates, such as those bearing a tert-butyl group, yield anti-Markovnikov products by placement of the terminal alkynyl H (rather than the bulkier alkynyl substituent) near the tert-butyl group to form a lower energy (and thus more stable) azametallocycle intermediate. With aryl amine substrates, this steric interaction can be avoided by strategic rotation of the aryl group to be perpendicular to the plane of the azametallocycle, and thus the Markovnikov product can be obtained.3
Lastly, catalysts containing group 5 metals such as V and Ta are known to catalyze hydroamination of alkynes although their efficiency is less than that of the group 4 metals. The catalytic cycles for these group 5 catalysts are thought to be the same as those for the group 4 metals, although there is current speculation to that regard.3
Allenes
Hydroamination of allenes follows the same catalytic cycle as hydroamination of alkynes (Scheme 14) with early transition metal catalysts. A significant difference in allene hydroamination is the ability to form various regioisomers, such as imines or allylamines, from the ability to form the azametallocycle from one of the two different double bonds. An illustrative example of intramolecular hydroamination of aminoallenes is provided in Scheme 15.
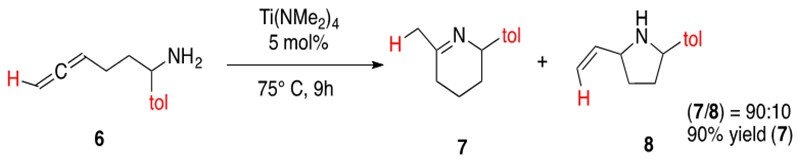
Scheme 15: Intramolecular hydroamination of an amionallene 6 yielding a regioisomeric mixture of products, imine 7 and allyl amine 8.3
Regioselective hydroamination of allenes has been reported with early transition metal catalysts. In general, monosubstituted aminoallenes will form preferentially the imine product and 1,3-disubstituted or trisubstituted aminoallenes will form preferentially the ally amine product. Allene hydroamination has also been performed intramolecularly and intermolecularly.3
Alkenes
Alkenes undergo early transition metal catalyzed hydroamination through two different mechanistic pathways: (1) the cationic catalyst pathway and (2) the neutral catalyst pathway. Secondary aminoalkenes require a cationic catalyst because they cannot form the catalytically active metal imido species. Primary aminoalkenes undergo the neutral catalyst pathway as they can from this metal imido species.
We will first consider the primary aminoalkene hydroamination with an example reaction (Scheme 16) followed by the mechanism (Scheme 17). This mechanism (neutral catalyst) resembles the mechanistic pathway of alkynes introduced in the beginning of this section.
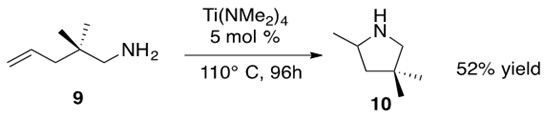
Scheme 16: Intramolecular hydroamination of a primary aminoalkene 9 to afford cyclic amine 10 in 52% yield.3
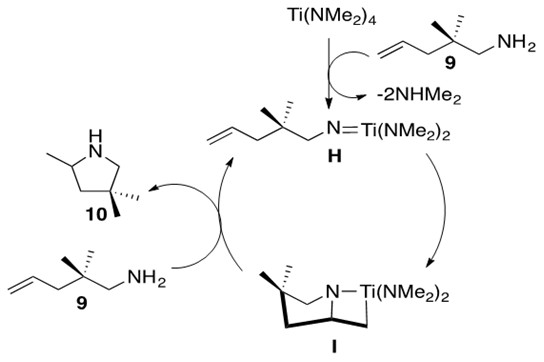
Scheme 17: The mechanism for the neutral early transition metal catalyst for hydroamination of a primary alkene.3
One feature of the mechanism depicted in Scheme 17 is the formation of the Ti imido species H from the Ti precatalyst. This species undergoes intramolecular [2+2] cycloaddition to form intermediate I, containing the familiar azametallocycle. Upon addition of another equivalent of aminoalkene 9, I undergoes a-elimination to afford product 10 and regenerate species H. It should be noted that other possible metals to perform this transformation include Zr and Hf.3
Hydroamination of secondary aminoalkenes cannot proceed by the mechanism presented above. Cationic metal catalysts, such as J in Figure 9, are employed to facilitate this reaction. An illustrative example of such a reaction is presented in Scheme 18.
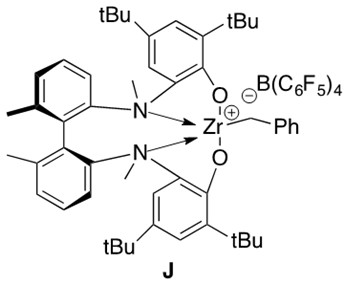
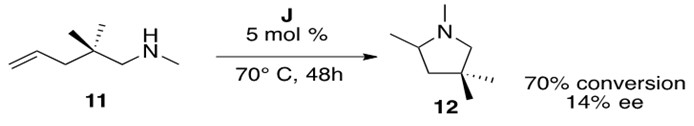
Scheme 18: Cationic zirconium catalyzed intramolecular hydroamination of a secondary aminoalkene 11 to form cyclic amine 12.
We will consider catalytic cycle for this hydroamination in Scheme 19 below:

Scheme 19: Cationic metal mechanism for hydroamination of a secondary amine, where the –[B(C6F5)4]- anion was removed for the sake of simplicity (but note that it should be present in all steps of the reaction to balance the charge on Zr).
The Zr catalyst undergoes a-elimination to yield the cationic Zr amido intermediate K. A proposed 4-membered transition state L yields the cyclic product bound to the cationic Zr intermediate M which upon a-elimination yields the catalytically active intermediate K and product 12. Note that the above mechanism resembles the mechanism presented for lanthanide-metal catalyzed hydroamination.
Application to Total Synthesis
The first natural product to be synthesized using early transition metal hydroamination was monomorine (15, Scheme 20), a pheromone found in monomorium pharaonis that causes ants to be lure ants.[5] McGrane and Livinghouse synthesized a titanium catalyst to convert a substituted alkyne 13 into a cyclic organotitanium compound 14, which was then used to synthesize the final product monomorine 15. Scheme 20 shows the synthetic pathway undertaken to produce monomorine. The mechanism proceeds through a [2+2] reaction, presented in Scheme 14.
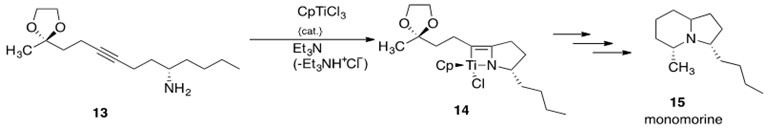
Scheme 20: The synthesis of the natural product monomorine 15, which utilizes early transition metal-catalyzed intramolecular hydroamination to convert alkyne 13 to organotitanium compound 14 in the synthetic sequence.5
LATE TRANSITION METAL CATALYSTS
Hydroamination catalysts derived from late transition metals are a chemically rich group. The d8/d10 metal species associated with these catalysts and their ability to act as Lewis Acids provide a high degree of activity toward hydroamination by varied mechanistic pathways. These robust catalysts generally demonstrate low reactivities with air and water and good functional group tolerance. In addition, these metals catalyze reactions which mainly proceed with Markovnikov regioselectivity.
Metals such as Ru, Rh, Pd, Ir, Pt, Ni, Cu, Zn, and Ag have all been found to catalyze hydroamination. Thus, this section deals with a group of catalysts that is too broad to consider by substrate scope as in the previous sections. Rather, we will consider four essential mechanistic pathways through which late transition metal catalyzed hydroamination is proposed to take place, providing example catalysts and reactions as well as considering substrate scope for each mechanistic pathway. The four mechanistic possibilities are divided into two groups, each of which features one of two key steps: (1) nucleophilic attack by nitrogen on a metal-coordinated, unsaturated C-C bond or (2) metal-hydride or metal-amide bond insertion by unsaturated C-C bond.
Nucleophilic Attack: Nitrogen on Coordinated Alkene/Alkyne
This mechanistic pathway is perhaps the most intuitive. It has been thoroughly studied and proposed for many different catalysts, including Ir, Pd, Pt, Cu, and Au. It requires alkene or alkyne as substrate and can occur intermolecularly or intramolecularly.
Consider the illustrative example reaction in Scheme 21.
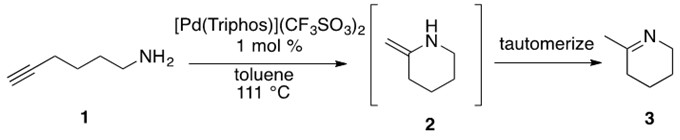
Scheme 21: Aminoalkyne 1 undergoes cationic Pd-catalyzed hydroamination to afford enamine 2 which rapidly tautomerizes to imine product 3.[6]
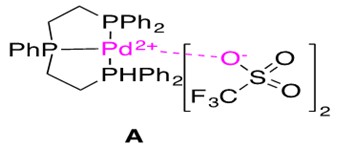
The catalyst used for this reaction is shown in Figure 10 and the proposed mechanistic pathway is presented in Scheme 22.
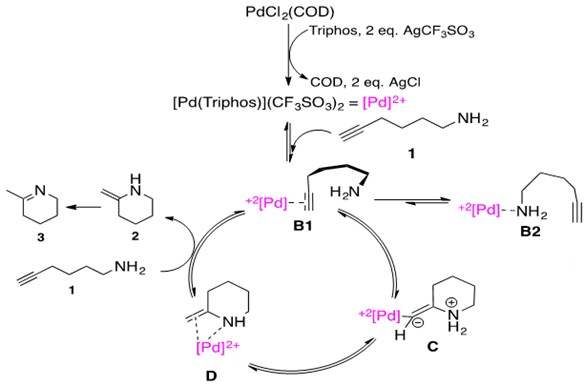
Scheme 22: Cationic Pd catalyzed intramolecular hydroamination of aminoalkyne via nucleophilic attack of nitrogen on a coordinated alkyne. Note two CF3SO3 anions have been omitted from species B1 – D for simplicity.3
Let us consider the mechanistic pathway in Scheme 22. The precatalyst PdCl2(COD) is treated with Triphos ligand and 2 equivalents of Brønstead base AgCF3SO3 to generate the catalytically active [Pd(Triphos)](CF3SO3)2 abbreviated as [Pd]2+ for simplicity. Notice the metal center maintains a (II) oxidation state throughout the catalytic cycle, characteristic of late transition metal catalyzed hydroamination reactions. Aminoalkyne substrate 1 first coordinates to the cationic, Lewis Acidic Pd species. This can happen by one of two ways: (1) through alkyne coordination, B1, or (2) through amine coordination, B2. In species B1, the coordination through alkyne thus activates this unit for intramolecular nucleophilic attack by the pendant nitrogen.
As a general principle, the nucleophile attacks at the internal position, allowing the bulkier Pd substituent to occupy the terminal end. This affords zwitterionic species C which undergoes protolytic cleavage to afford the enamine product 2 coordinated to the metal center, D. Upon addition of an equivalent of substrate 1, the enamine 2 dissociates. Given that the starting material is a primary amine, this material then rapidly tautomerizes to the imine 3.
The most important feature of this cycle is activation of the unsaturated C-C bond via metal coordination. Thus, the metal in the catalytic cycle must be able to serve as a Lewis Acid for this purpose. Additionally, Markovnikov addition products are expected for this cycle due to amine attack at the internal carbon. The rate-determining step was found by kinetic studies and DFT studies to be the protolytic cleavage of the Pd-C bond in intermediate C.3
We would like to highlight the use of a Brønstead acid co-catalyst in this reaction, which has been shown to improve the rate of the reaction. Although the role of co-catalytic acid is not fully understood, some possible explanations3 include:
(1) inhibition of amine coordination (B2) by forming ammonium salt
(2) assisting in the rate-determining protolytic cleavage step (C to D) as external proton source
(3) assisting in tautomerization of enamine 2 to imine 3 as external proton source, preventing the enamine from coordinating to metal catalyst which would impede catalytic activity.
All of these explanations seem reasonable and likely, yet the mechanism for the rate-determining protolytic cleavage is not fully understood.
The choice of counter ion in co-catalyst also affects the rates of these hydroamination reactions. It was found that bulkier anions tend to increase reaction rates (e.g., OTf- > BF4- > PF6- > NTf2-), likely as a function of their poorer ability to coordinate to the metal center and inhibit catalytic activity.3
From the acidic co-catalyst discussion, it is clear that interaction of amine with the Lewis Acidic metal center must be taken into consideration for these systems. Thus, choice of amine substrate is important in these reactions. More basic amines will poison the catalyst by preventing coordination of unsaturated C-C bonds to initiate the reaction. This problem is often addressed by using protected amines or less nucleophilic anilines, carboxamides, and sulfonamides.3
Choice of ligand also influences these reactions. Strongly binding tridentate ligands (such as Triphos, shown in Figure 10) improves the reaction by preventing b-H elimination in species B (given that the starting material is an alkene and not an alkyne – alkynic starting material will not possess b hydrogens). Therefore, although the effect of the tridentate ligand was not in play during this reaction, it is an important consideration and comment on catalyst design.3
As previously stated, the protolytic step of this reaction is not well understood. We will consider the Pt-mediated intermolecular hydroamination of alkene and arylamine to illustrate the nuances of late transition metal chemistry (Scheme 23).
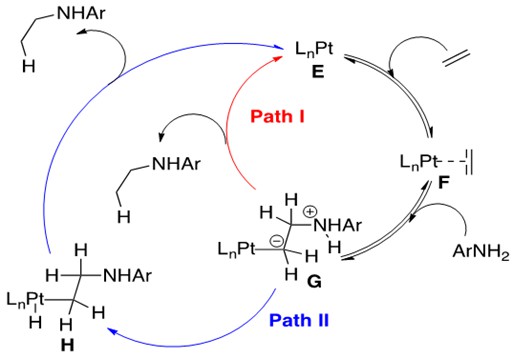
Scheme 23: Pt-mediated intermolecular hydroamination, where protonolysis of the Pt-C bond and release of product proceeds by two different pathways, Path I or Path II.3
Scheme 23 depicts the familiar first two steps of this reaction, olefin coordination to catalyst E to form F followed by nucleophilic attack on the coordinated olefin by amine to form G. It is here, however, that chemists consider two possible pathways for protonolysis and release of product. Path I, a one-step protonation of the a-carbon, causes release of product and returns the system to catalyst E. Alternatively, protonation of the metal center to form H followed by reductive elimination to release product and return to E is also feasible.
DFT experiments indicate that group 10 metals prefer Path I and group 9 metals prefer Path II.3 Thus variations within the same general catalytic cycle as functions of the metal center family are possible for hydroamination, illustrating the uniqueness of this group of metal catalysts.
Nucleophilic Attack: Nitrogen on Coordinated Allylic Species
The following mechanism is used to describe hydroamination of allenes, dienes, and trienes with catalysts such as Pd and Ni. By extension, it can be used to describe hydroamination of vinyl arenes and other molecules containing unsaturated C-C bonds in conjugation with another unsaturation that can support an h3-allyl structure.
We will begin by considering an illustrative example of this reaction in Scheme 24 followed by its proposed catalytic cycle in Scheme 25.
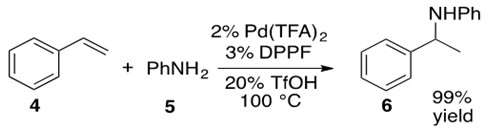
Scheme 24: Pd catalyzes the hydroamination of styrene 4 with aniline 5 to afford the N-phenyl phenylethyl amino product 6 in high yield.[7]
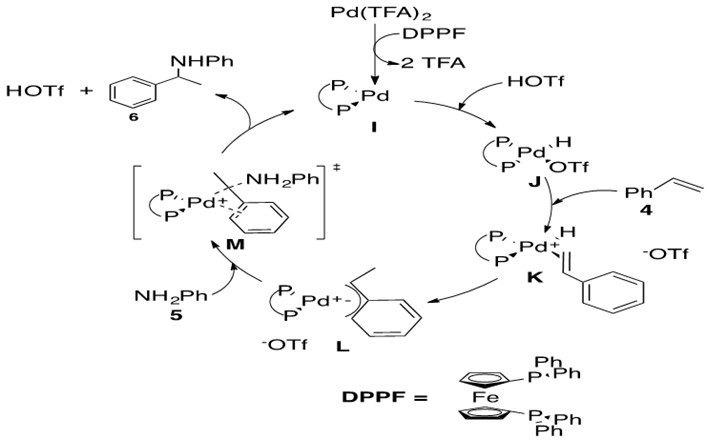
Scheme 25: Pd-catalyzed hydroamination via nucleophilic attack on an allylic intermediate.7
This reaction begins with formation of precatalyst into the catalytically active Pd(0) species I by introducing the bidentate phosphine ligand, DPPF. Metal-hydride J is formed via oxidative addition of HOTf. Styrene 4 coordinates via its terminal alkene to the Pd metal center, forming a cationic Pd intermediate K which is balanced with the OTf anion. b-Hydride insertion into the olefin leads to two possible regioisomers, one of which places the a-carbon in the allylic position of a double bond in the phenyl ring. This in turn leads to an h3-allyl bond to the Pd metal center, L. Aniline 5 nucleophilically attacks this allylic system opposite the side coordinated to the metal center in a trans-like fashion as shown in transition state M. Coordinated OTf anion deprotonates the ammonium species to afford Markovnikov product 6 which easily de-coordinates from the Pd center, yielding 6 and regenerating catalyst I.
The key feature of this mechanism is the formation of the h3-allylic species K. These species have been isolated and characterized by single crystal X-ray diffraction, indicating that they are lingering intermediates in the catalytic cycle. This suggests that the rate-determining (slow) step is nucleophilic attack of amine 5 on allylic species K. Kinetic isotope effect studies revealed that the regio-determining step is also the rate-determining step (nucleophilic attack of amine). It had been previously hypothesized that regioselectivity was determined by formation of the h3 intermediate L, but these studies revealed formation of h3 intermediate L is reversible. The rate of this reaction increases as the basicity of the amine increases and decreases with more sterically hindered amines, which more generally states the greater nucleophilicity of the amine, the greater the reaction rate.3
It should be noted for this reaction that use of stereochemically-enriched bidentate ligands such as BINAP affords enantioselective products.7 Thus hydroamination with this catalyst system can proceed enantioselectively.
There is an interesting debate regarding the order of the steps from species I to the allylic species L. The path shown above in Scheme 25 is oxidative addition of the acid across the metal, forming metal hydride J which then coordinates olefin, species K. An alternate path involves coordination of olefin first, followed by addition of acid generating allylic species. This latter pathway is proposed for hydroamination of trienes.3
Notice the above example proceeded with Markovnikov selectivity.3 Anti-Markovnikov selectivity has been reported in a Rh- and Ru-mediated hydroamination of vinyl arenes shown in Scheme 26.
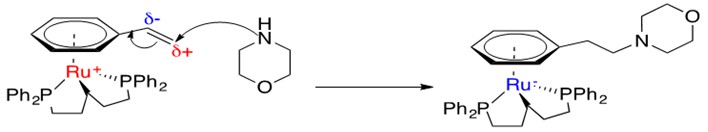
Scheme 26: Brief mechanistic explanation for observed anti-Markovnikov addition of amines across styrene with Ru catalysts.3
Ru is able to coordinate an h6-arene system with a pendant ethyl alkene group. Resonance donation into the ring allows for a partial cationic charge to develop on the terminal carbon, creating an electrophilic carbon and enabling anti-Markovnikov addition of the amine. The Ru metal center can absorb the negative charge that gets donated into the ring as a result of this addition; this forms a stable 18 e- species.3
Olefin Insertion into M-H Bond
We will revisit a previously mentioned reaction under a new set of conditions for a brief discussion of the lesser-known mechanistic pathway of olefin insertion into M-H bond. This mechanistic pathway applies mostly to alkene substrates and Pd catalysts.

Scheme 27: Heterogenous Pd-catalyzed hydroamination of styrene 4 with aniline 5 afforded anti-Markovnikov product 7 at high temperatures.[8]
The mechanism for Markovnikov product 6 was analyzed as part of the discussion of nucleophilic attack on metal coordinated-allylic species (Scheme 25). We will now consider a different mechanistic pathway that will afford anti-Markovnikov product, presented in Scheme 28 below.
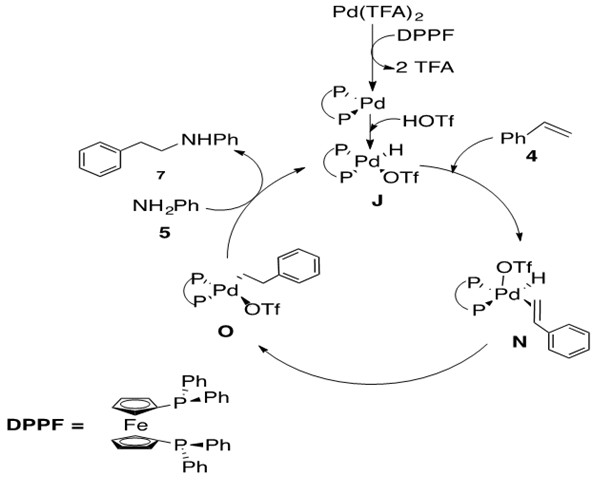
Scheme 28: Pd-catalyzed hydroamination of styrene with aniline to afford anti-Markovnikov product 7 via olefin insertion into Pd-H bond.8
The mechanism for Markovnikov and anti-Markovnikov product begin in similar ways. The difference occurs upon coordination of styrene 4 to catalytically active species J to afford the five-coordinate 18 e- Pd species N. b-hydride insertion to afford O followed by nucleophilic attack at the a-carbon of the alkyl chain releases product 7 and regenerates catalyst J.
The rate determining step was found to be the nucleophilic attack of amine, from O to J. Additionally regioselectivity was determined by the insertion step, from N to O, with insertion occurring preferentially at the carbon that would reduce steric interactions of the ligand with the alkenyl substituent (phenyl group). This explains the anti-Markovnikov selectivity for this reaction.8
Olefin Insertion into M-N Bond
This final mechanistic pathway resembles closely the pathway presented in (III), except the initial step is oxidative addition of an N-H bond of the amine substrate. This forms two s-bonds, M-N and M-H, of an hydrido-amido complex. The unsaturated C-C bond inserts itself preferentially into the M-N bond, as will be explained below. This mechanistic pathway has been proposed for many late transition metals, including Pd, Pt, Ir, and Ru. It applies to hydroamination of alkenes, alkynes, and vinyl arenes.
We will consider one example reaction (Scheme 29) and follow its catalytic cycle (Scheme 30).
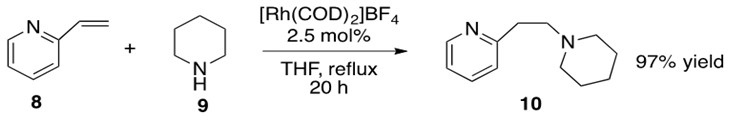
Scheme 29: 2-vinyl pyridine 8 undergoes hydroamination with piperidine 9 with Rh catalyst to yield anti-Markovnikov product 10 in high yield.9
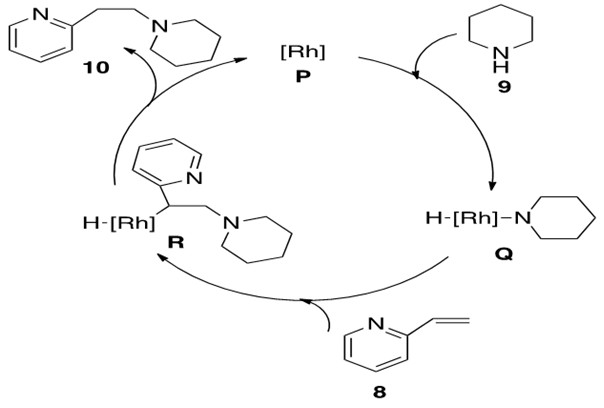
Scheme 30: The catalytic cycle for oxidative addition of piperidine 9 to Rh catalyst for hydroamination of alkene 8 to afford anti-Markovnikov product 10.[9]
The identity of the catalytically active [Rh] species P is not fully defined but is proposed to be a cationic 14 or 16 electron species that can undergo oxidative addition with amine 9. This yields the Rh-hydrido-amido complex Q which coordinates to alkene 8 and allows for b-insertion into the Rh-N bond, yielding Rh-alkyl-hydrido complex R. This complex readily reductively eliminates product 10 and reforms catalytically active species P.
The alkene could also have b-inserted into the Rh-H bond, but this is generally less favorable because reductive elimination of the resultant C-N bond to form product is more difficult to achieve than reductive elimination of C-H bond to form product.3
This reaction proceeded with anti-Markovnikov addition across the double bond. The explanation for regioselectivity in this mechanism appears to be sterics, but there is little experimental evidence to make this statement conclusive.8
DFT studies indicate that oxidative addition and reductive elimination were the rate-determining steps in a Pt-catalyzed hydroamination reaction that proceeded through this mechanism, although the kinetics for the reaction in Scheme 29 are not definitively known.3
Application to Total Synthesis
Secondary enamides are frequently found in biologically active molecules and are important synthons in natural product synthesis. Traditional organic chemistry affords secondary enamides via harsh conditions without stereoselectivity. Transition metal chemistry allows for an efficient method to stereoselectively synthesize these valuable substrates, thus overcoming a dilemma of traditional organic chemistry.
Gooßen’s Ru catalyst can synthesize the E or Z conformations as a function of added ligands.[10] Scheme 31 illustrates usage of Gooßen’s catalyst to afford the Z stereoisomer of 13, also known as lansiumamide A, a natural product.
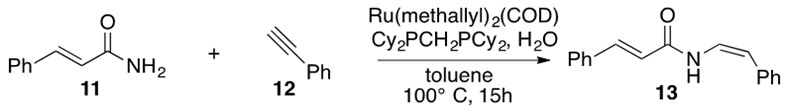
Scheme 31: Hydroamidation across alkyne 12 with amide 11 using Gooßen’s ruthenium catalyst with the appropriate ligands selected for the desired stereochemical outcome affords nautral product lansiumamide A 13.[11]
OTHER CATALYTIC METHODS
Although the scope of this paper is to address hydroamination through transition metal catalysts and organometallic chemistry, it would be negligent to study hydroamination without considering catalytic methods outside of organometallic chemistry.
Hydroamination can be accomplished through use of acid catalysts. An example is provided in Scheme 32 below.

Scheme 32: Hydroamination of tosylamine 2 over styrene 1 is catalyzed by HOTf in toluene at elevated temperature and sufficient reaction time to yield Markovnikov product 3.3
Additionally, alkali metals such as Na and Li can catalyze hydroamination. Consider the example reaction below.
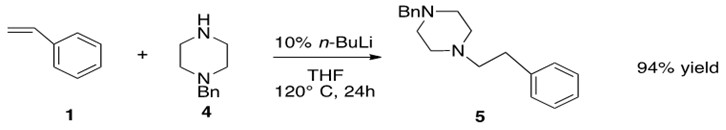
Scheme 33: Hydroamination of amine 4 with styrene 1 through assistance of n-BuLi to yield anti-Markovnikov product 5 in good yield.3
These are classic reactions taught in introductory organic chemistry classes. They efficiently produce the products of hydroamination that are attained through transition metal chemistry. They often require functionalization of the starting materials to obtain high yields. There are also many opportunities for functional group incompatibility, usually at the expense of yield.
We will very briefly consider the extensive field of mercury facilitated hydroamination. An example reaction is provided below (Scheme 34).
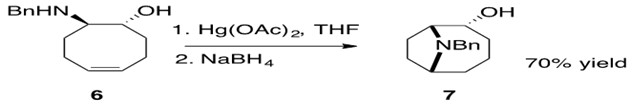
Scheme 34: Mercury facilitated intramolecular hydroamination of an aminoalkene.3
These generally efficient reactions proceed via a three-membered mercuronium, Hg(II), intermediate. One major drawback to these reactions is the toxicity of mercury.3 For applications in total synthesis and the pharmaceutical industry, hydroamination reactions must be consistent and predictable (in terms of regioselectivity, stereoselectivity), afford good yields, proceed at reasonable reaction times and temperatures, and be suitable for human consumption after product purification. Although mercury facilitated hydroamination is well studied, the latter restriction on the reaction’s usefulness in industry puts it at a clear disadvantage. The various transition metal catalysts discussed in sections 1 to 3 fulfill these criteria most effectively, as shown in the total synthesis examples provided at the end of each section.
SUMMARY
This report dealt with catalytic organometallic hydroamination reactions. The first group of catalysts considered were those composed of the Lanthanide series of metals, which catalyze hydroamination of alkenes, alkynes, allenes, and dienes. The catalytic cycle for these reactions involves a 4-membered transition state that forms an azacylce, that can be either vinylic to or allylic to the metal center. These reactions result in a cis addition of amine and hydrogen across the unsaturated C-C bond. Lanthanide metal catalyzed reactions are usually restricted to intramolecular reactions, which leads to inner-sphere attack of the nitrogen on the unsaturated C-C bond and affords cis product.
The second group of catalysts considered were the Early Transition Metals. These catalysts require formation of an M=N (imido) or M-N (amido) species that undergoes a [2+2] cycloaddition mechanism to afford a azametallocycle. There are both intermolecular and intramolecular examples of hydroamination afforded by these catalysts for alkenes, alkynes, and allenes. Thus outer-sphere attack can occur, opening up the possibility for formation of trans product.
Lastly we considered the Late Transition Metals, which offer a broad range of mechanistic possibilities. Both intermolecular and intramolecular examples of these reactions exist, and they generally occur through use of co-catalytic acid. In general, the mechanism is decided based on whether nitrogen attacks the C-C p-bond or whether the C-C p-bond inserts (“attacks”) the M-N bond. There are also examples of the C-C p-bond inserting into M-H bonds, although this is a less favorable process than inserting into M-N bonds. Late Transition Metals catalyze hydroamination across alkenes, alkynes, dienes, and allenes. Examples of anti-Markovnikov addition are abundant in Early Transition Metal and Late Transition Metal catalyzed reactions, and Markovnikov addition is found often in all catalyst groups. Enantioselectivity has been reported through use of chiral ligands. There is still much to be understood about hydroamination chemistry, but its offerings to synthetic organic chemists thus far render it a field of valuable organometallic research.
Table 1 concludes this report and provides the student of organometallic chemistry with an easy reference guide for the information presented above. The table is organized by catalyst type, which constitutes the three main sections of this report, a general assignment of the oxidation state of the metal center throughout each catalytic cycle, schemes for relevant catalytic cycles for further reference, key features of each catalytic cycle, and factors that have been reported to affect regioselectivity.
Table 1: A general breakdown of the content in this report categorized by the three main sections of transition metal (TM) catalyst types (Lanthanide, Early TM, and Late TM), the general oxidation state of the metal during the catalytic cycle, pertinent mechanistic schemes for each general catalytic cycle, key features of each mechanism, and factors that determine regioselectivity discussed in this report.
|
Catalyst Type |
Metal Center Oxidation State |
Catalytic Cycles |
Key Features |
Regioselectivity (Markovnikov vs. anti-Markovnikov) |
|
Lanthanide |
(III) |
Scheme 4 |
*Azametallocycle transition state/transient species *Azacycle intermediate |
*Electronic effects govern addition |
|
Early TM |
(IV) |
Schemes 14, 19 |
*Primary amines form metal imido (M=N) species *Secondary amines form metal amido (M-N) species *Azametallocycle intermediate |
*Steric effects govern addition |
|
Late TM |
(II) |
Schemes 22, 24, 28, 30 |
*Varied mechanistic schemes *Often with co-catalytic acid *Occur via nucleophilic attack by amine or olefin insertion * C-C p -bond coordinates to metal first (for all but Scheme 30) *Oxidative addition of amine first (Scheme 30) |
*Steric effects govern addition |
REFERENCES
[1] Hong, S.; Marks, T.J. Acc. Chem. Res. 2004, 37, 673-686.
[2] Yuen, H.F.; Marks, T.J. Organometallics, 2009, 28(8). 2423–2440.
[3] Müller, T.E.; Hultzsch, K.C.; Yus, M.; Foubelo, F.; Tada, M. Chem. Rev. 2008, 108, 3795-3892.
[4] Müller, T.E.; Beller, M. Chem. Rev. 1998, 98(2), 675-703.
[5] McGrane, P.L.; Livinghouse, T. J. Org. Chem. 1992, 57(5), 1323-1324.
[6] Penizen, J.; Su, R.Q.; Müller, T.E. J. Mol. Cat. A: Chem. 2002, 182-183, 489-498.
[7] Vo, L.K.; Singleton, D.A. Org. Lett., 2004, 6(14), 2469-2472.
[8] Sievers, C.; Jiménez, O.; Knapp, R.; Lin, X.; Müller, T.E.; Türler, A.; Wierczinski, B.; Lercher, J.A. J. Mol. Cat. A.: Chem. 2008, 279, 187-199.
[9] Beller, M.; Trauthwein, H.; Eichberger, M.; Breindl, C.; Müller, T.E. Eur. J. Inorg. Chem. 1999, 1121-1132.
[10] Gooßen, L.J.; Rauhaus, J.E.; Deng, G. Angew. Chem. Int. Ed. 2005, 44, 4042-4045.
[11] Gooßen, L.J.; Blanchot, M.; Arndt, M.; Kifah, S.M. Synlett. 2010, 11, 1685-168
Contributors and Attributions
- Alec Beaton and Victoria Banas