
Stille Coupling
- Page ID
- 69098
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)The Stille reaction, named after the late John Kenneth Stille, is a palladium-catalyzed cross coupling reaction. Heavily used in organic synthesis, it involves the coupling of an organic halide with an organotin compound.1,2 The reaction proceeds according to the following equation11:
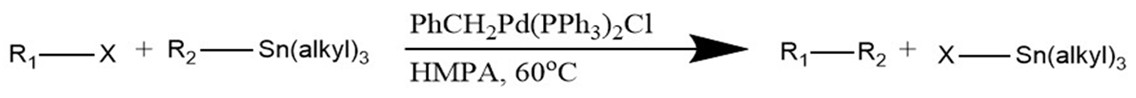
General Stille Coupling Reaction
The reaction proceeds using a palladium phosphine catalyst. In this reaction, R1 and R2 represent the organic chains that are to be coupled. These are usually chains containing an sp2 hybridized carbon (aryl, alkenyl, allyl.) X represents a leaving group such as a halide or triflate (Cl, Br, I, OTf, etc.) In some instances, it is possible to use sp3 hybridized alkyls and aromatics as R1. It’s worth noting that, although air stable, tin reagents tend to be highly toxic.2
History
Prior to Stille’s work, palladium-catalyzed cross coupling reactions had been observed. The foundations were laid by Eaborn and Migita in the late 70’s. Eaborn’s process involved formation of diary product using an organotin reagent. This was expanded by Migita who coupled acyl chlorides with organotin reagents to form ketones with yields ranging from 53-87%.3
Later on, Migita was able to couple aryl and acyl halides with allyl tin reagents. He was able to do so at lower temperatures due to allyl’s ability to interact more efficiently with the palladium catalyst. Yields for both aryl and acyl halides were inconsistent ranging from as little as 4% to as much as 100%.4
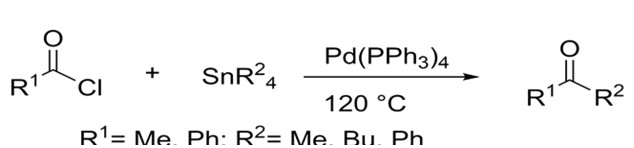
Migita succeeded in coupling acyl chlorides to organotin reagents under harsh conditions4
Stille was able to build upon these foundations using a variety of alkyl tin reagents. In 1978, he published a report on the successful coupling of many alkyl tin reagents with myriad acyl and aryl halides. He was able to carry out these reactions under much more mild conditions and obtaining much higher yields (76-99%). Subsequent research focused on synthesizing a variety of compounds in this fashion.5,6
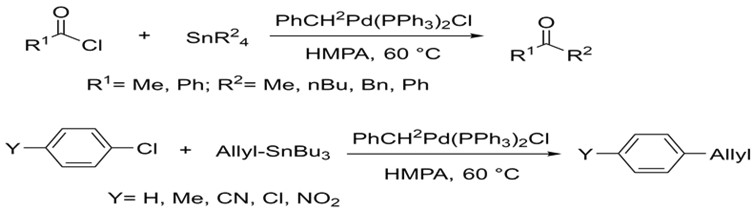
Stille's research focused on coupling many organic halides to organotin reagents using palladium under milder conditions.5,6
Scope & Limitations
Stille coupling is a commonly used procedure because of its wide scope. There are ample choices for both nucleophile and electrophile. Furthermore, organotin reagents are air stable, commercially available or readily synthesized, making Stille coupling an easily accessible method for synthesis. A wide variety of aryl, vinyl and acyl halides or pseudo halides can be used as electrophiles. Furthermore, heterocyclic compounds are also viable choices.12The example below shows that the Stille reaction can also work with purines to build more complex molecules.
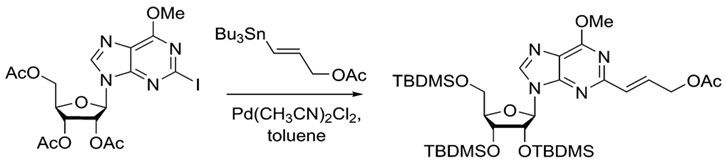
.
The Stille reaction can add complexity to purines
The major exceptions to this are organic chlorides which are not reactive enough to undergo oxidative addition with the palladium catalyst. As a result, organic bromides or iodides are preferred.
Similarly, the organotin reagent shows great versatility. As with the electrophile, many classes of organotin reagents work well in Stille coupling. Aryl, alkyl, vinyl and even heterocyclic stannanes are known to work. The following example shows N, S containing heterocyclic ring participating in Stille coupling.7
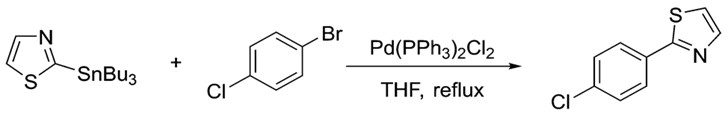
Heterocyclic compounds are capable of participating in Stille coupling; showing the versatility of the reaction.
Limitations include very bulky or heavily substituted reagents. These tend to react very slowly and may require optimization, typically in the form of co-catalytic copper iodide. Nonetheless, the Stille reaction is a versatile one of great importance to organic synthesis.
Stereo & Regioselectivity
Stereochemistry is typically retained in Stille coupling. Regioselectivity, as with most other coupling reactions is hard to control though optimizations have been found to control regioselectivity to some extent. Despite advances by researchers in controlling stereoselectivity, mechanisms have not been fully elucidated and require further study. 7,8,10
Synthetic Applications
The synthetic applications of Stille coupling are immense. A reaction of this versatility and scope readily found its way into total syntheses of natural products. Stille coupling has been of great use towards the synthesis of many such products, many of these antibiotics or anticancer drugs. Among these are manzamine A, ircinal A, oxazolomycin and many others. 9
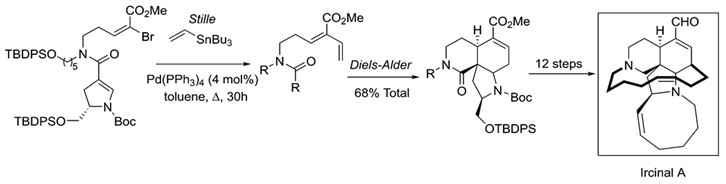
Stille coupling is an important step in the total synthesis of the marine alkaloid Ircinal A. Ircinal A is a known antitumor substance.9
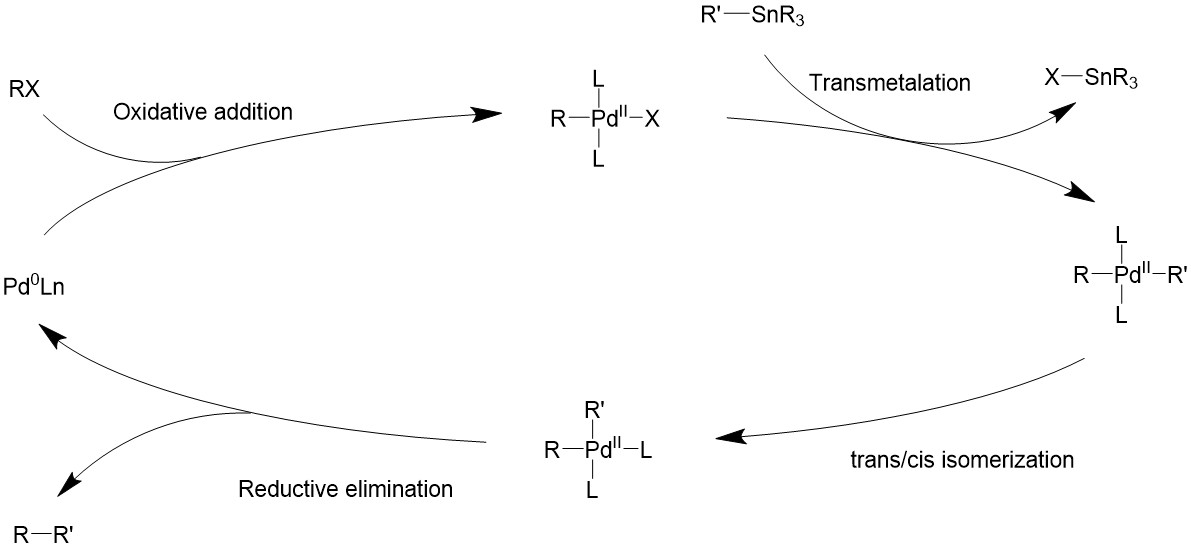
Mechanism
Catalyst design
The Stille reaction uses a palladium catalyst. It can use an 18- or 16-electron Pd (0) complex as a source of the catalyst, such as Pd(PPh3)4, Pd(dba)2. Then through ligand dissociation, it can be formed into a 14-electron Pd (0), the active catalyst.
Upon oxidative addition, the electrophile will bind to palladium, turning it into a 16-electron square planer intermediate. The organostannane then comes in as a transmetalation agent to introduce the R’ group and also taking away the halide. Finally, the two R groups will be coupled by reductive elimination through various possible pathways, and turning the catalyst back to the 14-electron Pd (0).
Catalytic cycle
Oxidative addition
The alkyl halide would first perform oxidative addition to the palladium in a concerted fashion, resulting in a 16-electron Pd(II) intermediate. The cis square planer product is in a fast equilibrium with the trans product.The bulky ligands used on the catalyst makes the trans product more thermodynamically stable, therefore most of the intermediate will isomerize into the trans product15,16.
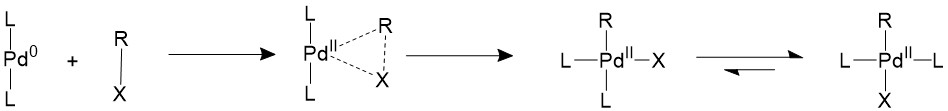
A trans- intermediate can also be formed when the catalyst reacts with an sp3 organohalide in a SN2 mechanism.
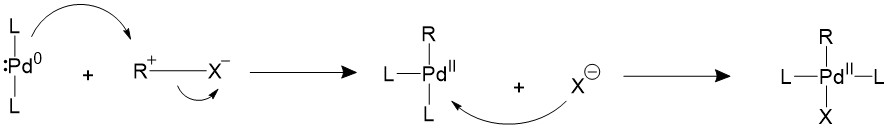
Transmetalation
The Stille coupling uses organostannane as a trans coupling reagent. The tin is usually bound to allyl, alkenyl, or aryl groups. The tin and the R’ group will form a four-member ring with the palladium center and the halide, forming an 18-electron transition state. Then the tin halide will leave and the R’ group stays bonded to palladium17.
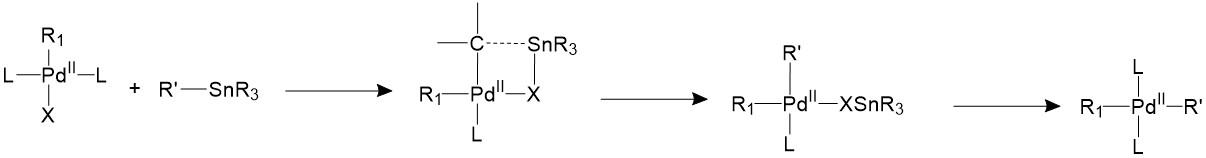
Reductive elimination
The two trans- R groups must first isomerize back into a cis- conformation, then it can undergo a concerted reductive elimination17-19.
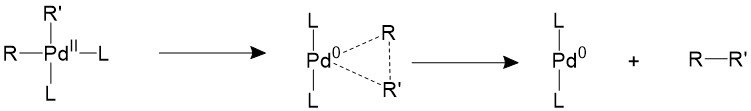
There are other two proposed mechanisms which involves the association or dissociation of the ligands. An extra ligand can be bond to palladium, forming an 18-electron trigonal bipyramidal structure and forces the two R groups to the equatorial position, which is a suitable conformation for them to form C-C bonds.
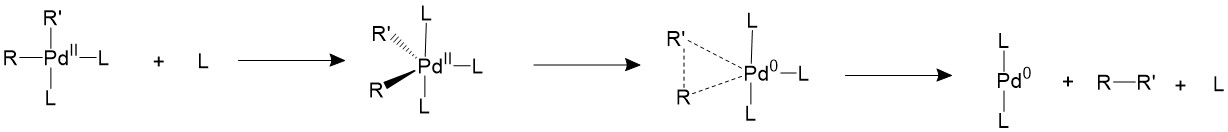
Another proposed mechanism is that one of the ligands will dissociate first to form a T-shape 14-electron intermediate, which is able to speed up the reductive elimination process.
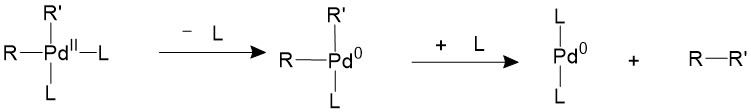
If the ligands are bulky enough, like phosphines with large cone angles, it is possible to push the two R groups closer to each other into an appropriate coordination angle, thus speeding up the reductive elimination process11.
Ligands
Various ligands are used as the 16- or 18- electron Pd0 catalyst, such as Pd(PPh3)4 and Pd(dba)213.

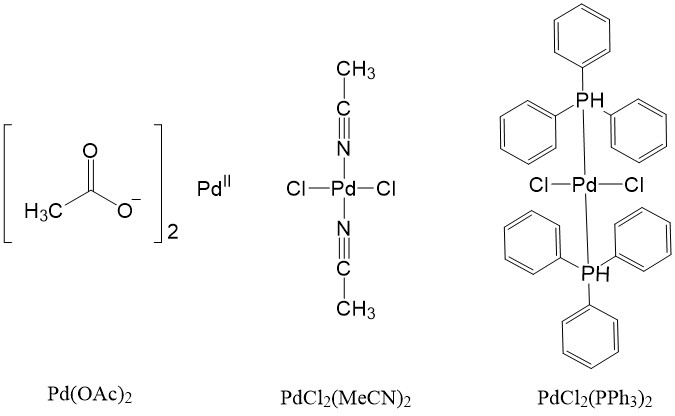
Some Pd(II) complexes can also be used as a source and can be reduced to the active Pd(0) catalyst, such as Pd(OAc)2, PdCl2(MeCN)2, PdCl2(PPh3)2.
Organostannane
Methyl and butyl group are often used as the other three R groups that binds to stannane, although these alkyl groups are not as reactive in the transmetalation process, there is still possibility that the alkyl coupling side product would form. This is called a” Non-transferable” ligand product. Though there are cases that shows benzylstannane are able to couple through Stille catalysis at higher temperatures. Both electron donating and electron withdrawing properties of the R’ group is favorable for transmetalation. The reactivity of the R group on organostannane have the following order20:
Alkynyl > Alkenyl > Aryl > Allyl = Benzyl > α-alkoxyl > Alkyl
Electrophiles
Vinyl halides, aryl halides and heterocyclic halides can all be used as the electrophile for the reaction. Iodine and bromide are used for the halogen because the chlorides are too inert for oxidative addition, and iodides would react even faster than bromide, as can be seen from the reaction below7:
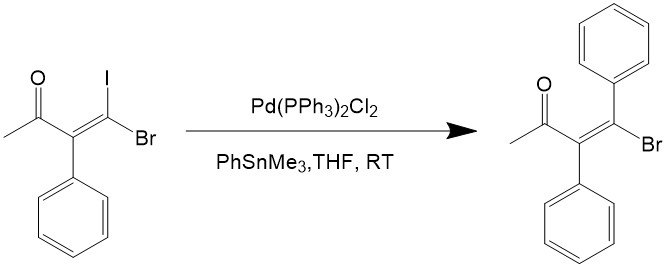
The stereochemistry is often retained during the catalytic reaction, unless it’s carried out under a harsh condition, so the Stille coupling reaction is quite good towards steroselectivity.
Additives
LiCl is often used to enhance the reaction rate by stabilizing the transition state during oxidative addition. It can also improve the rate of transmetalation by increasing the solvent’s polarity2,7. In addition, Cu(I) and Mn(II) salts are also used to increase reaction rate as well as improving selectivity.
References
- Robert W Lenz. In memory of John Kenneth Stille. Macromolecules, 1990, 23, 2417-2418
- Stille, J. K., The Palladium-Catalyzed Cross-Coupling Reactions of Organotin Reagents with Organic Electrophiles [New Synthetic Methods (58)]. Angew. Chem. Int. Ed. 1986, 25: 508–524.
- Azarian, D.; Dua, S. S.; Eaborn, C.; Walton, D. R. M. Reactions of organic halides with R3 MMR3 compounds (M = Si, Ge, Sn) in the presence of tetrakis(triarylphosphine)palladium. J. Organomet. Chem., 1976, 117, C55-C57.
- Kosugi, M.; Shimizu, Y.; Migita. Reaction of allyltin compounds: II. Facile preparation of allyl ketones via allyltins. T. Chem. Lett., 1977, 6, 1423-1424.
- Milstein, D.; Stille, J. K. Palladium-catalyzed coupling of tetraorganotin compounds with aryl and benzyl halides. Synthetic utility and mechanism. J. Am. Chem. Soc., 1979, 101, 4992-4998.
- Milstein, D.; Stille. Mild, selective, general method of ketone synthesis from acid chlorides and organotin compounds catalyzed by palladium. J. K. J. Org. Chem., 1979, 44, 1613-1618.
- Farina, V.; Krishnamurthy, V.; Scott, W. J. ‘‘The Stille Reaction’’; Wiley: Online, 2004.
- Farina, V. New perspectives in the cross-coupling reactions of organostannanes. Pure & Appl. Chem., 1996, 68, 73-78.
- Martin, S. F.; Humphrey, J. M.; Ali, A.; Hillier, M. C. Enantioselective Total Syntheses of Ircinal A and Related Manzamine Alkaloids. J. Am. Chem. Soc., 1999, 121, 866-867.
- Won-Suk Kim, Hyung-Jin Kim, and Cheon-Gyu Cho. Regioselectivity in the Stille Coupling Reactions of 3,5-Dibromo-2-pyrone. J. Am. Chem. Soc., 2003, 125 (47).
- Crabtree, R. H. The Organometallic Chemistry of the Transition Metals, 5th ed.; Wiley: New York, 2009.
- Vasu Nair, Gregory A. Turner, and Stanley D. Chamberlain. Novel approaches to functionalized nucleosides via palladium-catalyzed cross coupling with organostannanes. J. Am. Chem. Soc.1987 109 (23)
- Kurti, L.; Czako, B. ‘‘Strategic Applications of Named Reactions in Organic Synthesis’’; Elsevier: Burlington, 2005.
- Vittorio Farina, Bala Krishnan. Large rate accelerations in the stille reaction with tri-2-furylphosphine and triphenylarsine as palladium ligands: mechanistic and synthetic implications. J. Am. Chem. Soc., 1991, 113 (25), pp 9585–9595
- Espinet, P.; Echavarren, A. M. The Mechanisms of the Stille Reaction. Angew. Chem. Int. Ed., 2004, 43, 4704-4734.
- Perez-Temprano, M. H.; Gallego, A. M.; Casares, J. A.; Espinet, P. Stille Coupling of Alkynyl Stannane and Aryl Iodide, a Many-Pathways Reaction: The Importance of Isomerization. Organometallics, 2011, 30, 611-617.
- Garcia-Melchor, M.; Braga, A. A. C.; Lledos, A.; Ujaque, G.; Maseras, F. Computational Perspective on Pd-Catalyzed C–C Cross-Coupling Reaction Mechanisms. Acc. Chem. Res., 2013, 46, 2626-2634.
- Gillie, A.; Stille, J. K. Mechanisms of 1,1-reductive elimination from palladium. J. Am. Chem. Soc., 1980, 102, 4933-4941.
- Brown, J. M.; Cooley. Carbon-carbon bond formation through organometallic elimination reactions. N. A. Chem. Rev., 1988, 88, 1031-1046.
- McKillop, A.; Abel, E. W.; Stone, F. G. A.; Wilkinson, G. Comprehensive Organometallic Chemistry II, Elsevier Scientific: Oxford, 1995.
Contributors and Attributions
- Kevin Maldonado and Jeffrey Liu