
Raman: Application
- Page ID
- 1849
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)If one can extract all of the vibrational information corresponds a molecule, its molecular structure can then be determined. In the field of spectroscopy, two main techniques are applied in order to detect molecular vibrational motions: Infrared spectroscopy (IR) and Raman spectroscopy. Raman Spectroscopy has its unique properties which have been used very commonly and widely in Inorganic, Organic, Biological systems [1] and Material Science [2], [3], etc.
Introduction
Generally speaking, vibrational and rotational motions are unique for every molecule. The uniqueness to molecules are in analogous to fingerprint identification of people hence the term molecular fingerprint. Study the nature of molecular vibration and rotation is particularly important in structure identification and molecular dynamics. Two of the most important techniques in studying vibration/rotation information are IR spectroscopy and Raman spectroscopy. IR is an absorption spectroscopy which measures the transmitted light. Coupling with other techniques, such as Fourier Transform, IR has been highly successful in both organic and inorganic chemistry.
Unlike IR, Raman spectroscopy measures the scattered light (Figure 2). There are three types of scattered lights: Rayleigh scattering, Stokes scattering, and anti-stokes scattering. Rayleigh scattering is elastic scattering where there is no energy exchange between the incident light and the molecule. Stokes scattering happens when there is an energy absorption from the incident light, while anti-stokes scattering happens when the molecule emites energy to the incident light. Thus, Stokes scattering results in a red shift, while anti-stokes scattering results in a blue shift. (Figure 1) Stokes and Anti-Stokes scattering are called Raman scattering which can provide the vibration/rotation information
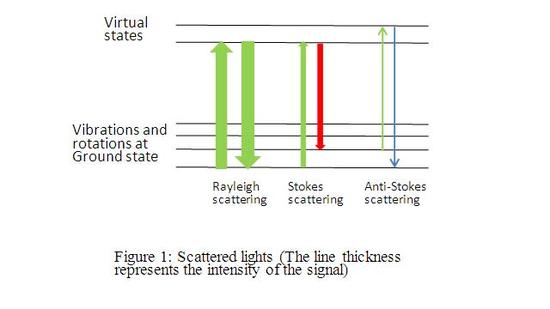
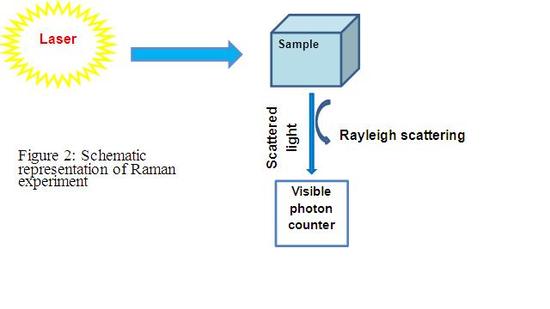
The intensity of Rayleigh scattering is around 107 times that of Stokes scattering. [4]According to the Boltzmann distribution, anti-Stokes is weaker than Stokes scattering. Thus, the main difficulty of Raman spectroscopy is to detect the Raman scattering by filtering out the strong Rayleigh scattering. In order to reduce the intensity of the Rayleigh scattering, multiple monochromators are applied to selectively transmit the needed wave range. An alternative way is to use Rayleigh filters. There are many types of Rayleigh filters. One common way to filter the Rayleigh light is by interference.
Because of the weakness of Raman scattering, the resolving power of a Raman spectrometer is much higher than an IR specctrometer. A resolution of 105 is needed in Raman while 103 is sufficient in IR. [5] In order to achieve high resolving power, prisms, grating spectrometers or interferometers are applied in Raman instruments.
Despite the limitations above, Raman spectroscopy has some advantages over IR spectroscopy as follows:
- Raman Spectroscopy can be used in aqueous solutions (while water can absorb the infrared light strongly and affect the IR spectrum).
- Because of the different selection rules, vibrations inactive in IR spectroscopy may be seen in Raman spectroscopy. This helps to complement IR spectroscopy.
- There is no destruction to the sample in Raman Spectroscopy. In IR spectroscopy, samples need to disperse in transparent matrix. For example grind the sample in solid KBr. In RS, no such destructions are needed.
- Glass vials can be used in RS (this should only work in the visible region. If in UV region, glass is not applicable because it can strongly absorb light too.)
- Raman Spectroscopy needs relative short time. So we can do Raman Spectroscopy detection very quickly.
After analysis of the advantages and disadvantages of Raman Spectroscopy technique, we can begin to consider the application of Raman Spectroscopy in inorganic, organic, biological systems and Material Science, etc.
Applications
Raman Spectroscopy application in inorganic systems
X-ray diffraction (XRD) has been developed into a standard method of determining structure of solids in inorganic systems. Compared to XRD, it is usually necessary to obtain other information (NMR, electron diffraction, or UV-Visible) besides vibrational information from IR/Raman in order to elucidate the structure. Nevertheless, vibrational spectroscopy still plays an important role in inorganic systems. For example, some small reactive molecules only exist in gas phase and XRD can only be applied for solid state. Also, XRD cannot distinguish between the following bonds: –CN vs. –NC, –OCN vs. –NCO,–CNO vs. –ONC, -SCN vs. –NCS. [7] Furthermore, IR and Raman are fast and simple analytical method, and are commonly used for the first approximation analysis of an unknown compound.
Raman spectroscopy has considerable advantages over IR in inorganic systems due to two reasons. First, since the laser beam used in RS and the Raman-scattered light are both in the visible region, glass (Pyrex) tubes can be used in RS. On the other hand, glass absorbs infrared radiation and cannot be used in IR. However, some glass tubes, which contain rare earth salts, will gives rises to fluorescence or spikes. Thus, using of glass tubes in RS still need to be careful. Secondly, since water is a very weak Raman scatter but has a very broad signal in IR, aqueous solution can be directly analyzed using RS.
Raman Spectroscopy and IR have different selection rules. RS detects the polarizability change of a molecule, while IR detects the dipole momentum change of a molecule. Principle about the RS and IR can be found at Chemwiki Infrared Theory and Raman Theory. Thus, some vibration modes that are active in Raman may not be active IR, vice versa. As a result, both of Raman and IR spectrum are provided in the stucture study. As an example, in the study of Xenon Tetrafluoride. There are 3 strong bands in IR and solid Raman shows 2 strong bands and 2 weaker bands. These information indicates that Xenon Tetrafluoride is a planar molecule and has a symmetry of D4h. [8] Another example is the application of Raman Spectroscopy in homonuclear diatomic molecules. Homonuclear diatomic molecules are all IR inactive, fortunately, the vibration modes for all the homonuclear diatomic molecules are always Raman Spectroscopy active.
Raman Spectroscopy Application in Organic Systems
Unlike inorganic compounds, organic compounds have less elements mainly carbons, hydrogens and oxygens. And only a certain function groups are expected in organic specturm. Thus, Raman and IR spectroscopy are widely used in organic systems. Characteristic vibrations of many organic compounds both in Raman and IR are widely studied and summarized in many literature. [5] Qualitative analysis of organic compounds can be done base on the characteristic vibrations table.
| Vibration | Region(cm-1) | Raman intensity | IR intensity |
|---|---|---|---|
| v(O-H) | 3650~3000 | weak | strong |
| v(N-H) | 3500~3300 | medium | medium |
| v(C=O) | 1820~1680 | strong~weak | very strong |
| v(C=C) | 1900~1500 | very strong~medium | 0~weak |
“RS is similar to IR in that they have regions that are useful for functional group detection and fingerprint regions that permit the identification of specific compounds.”[1] While from the different selection rules of Raman Spectroscopy and IR, we can get the Mutual Exclusion rule [5], which says that for a molecule with a center of symmetry, no mode can be both IR and Raman Spectroscopy active. So, if we find a strong bond which is both IR and Raman Spectroscopy active, the molecule doesn't have a center of symmetry.
Non-classical Raman Spectroscopy
Although classical Raman Spectroscopy has been successfully applied in chemistry, this technique has some major limitations as follows[5]:
- The probability for photon to undergo Raman Scattering is much lower than that of Rayleigh scattering, which causes low sensitivity of Raman Spectroscopy technique. Thus, for low concentration samples, we have to choose other kinds of techniques.
- For some samples which are very easily to generate fluorescence, the fluorescence signal may totally obscure the Raman signal. We should consider the competition between the Raman Scattering and fluorescence.
- In some point groups, such as C6 , D6 , D6h , C4h , D2h, there are some vibrational modes that is neither Raman or IR active.
- The resolution of the classical Raman Spectroscopy is limited by the resolution of the monochromator.
In order to overcome the limitations, special techniques are used to modify the classical Raman Spectroscopy. These non-classical Raman Spectroscopy includes: Resonance Raman Spectroscopy, surface enhanced Raman Spectroscopy, and nonlinear coherent Raman techniques, such as hyper Raman spectroscopy
Resonance Raman Scattering (RRS)
The resonance effect is observed when the photon energy of the exciting laser beam is equal to the energy of the allowed electronic transition. Since only the allowed transition is affected, (in terms of group theory, these are the totally symmetric vibrational ones.), only a few Raman bands are enhanced (by a factor of 106). As a result, RRS can increase the resolution of the classical Raman Spectroscopy, which makes the detection of dilution solution possible (concentrations as low as 10-3 M). RRS is extensively used for biological molecules because of its ability to selectively study the local environment. As an example, the Resonance Raman labels are used to study the biologically active sites on the bond ligand. RRS can also be used to study the electronic excited state. For example, the excitation profile which is the Raman intensity as a function of incident laser intensity can tell the interaction between the electronic states and the vibrational modes. Also, it can be used to measure the atomic displacement between the ground state and the excited state.
Surface Enhanced Raman Scattering (SERS)
At 1974, Fleischmann discovered that pyridine adsorbed onto silver electrodes showed enhanced Raman signals. This phenomenon is now called surface enhanced Raman Scattering (SERS). Although the mechanism of SERS is not yet fully understood, it is believed to result from an enhancement either of transition polarizability, α,or the electric field, E, by the interaction with the rough metallic support.
Unlike RRS, SERS enhances every band in the Raman spectrum and has a high sensitivity. Due to the high enhancement (by a factor of 1010~11), the SERS results in a rich spectrum and is an ideal tool for trace analysis and in situ study of interfacial process. Also, it is a better tool to study highly diluted solutions. A concentration of 4x10-12 M was reported by Kneipp using SERS. [5]
Nonlinear Raman Spectroscopy
In a nonlinear process, the output is not linearly proportional to its input. This happens when the perturbation become large enough that the response to the perturbation doesn’t follows the perturbation’s magnitude. Nonlinear Raman Spectroscopy includes: Hyper Raman spectroscopy, coherent anti-Stokes Raman Spectroscopy, coherent Stokes Raman spectroscopy, stimulated Raman gain and inverse Ramen spectroscopy. Nonlinear Raman spectroscopy is more sensitive than classical Raman spectroscopy and can effectively reduce/remove the influence of fluorescence. The following paragraph will focus on the most useful nonlinear Raman spectroscopy---coherent anti-Stokes Raman Spectroscopy (CARS):
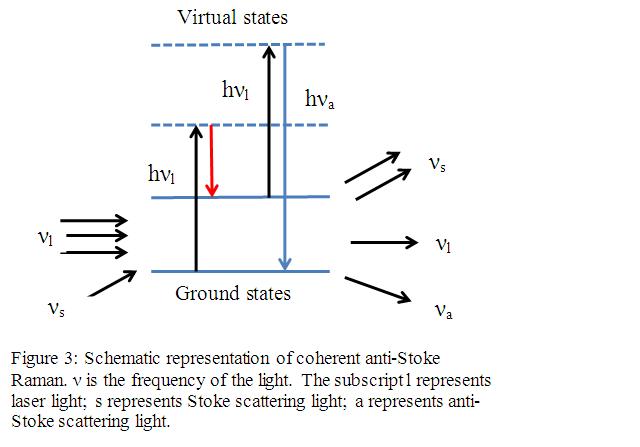
References
- Principles of Instrumental Analysis, fifth edition. Skoog, Holler and Nieman.
- Infrared and Raman Spectra of Inorganic and Coordination Compounds, fifth edition. Kazuo Nakamoto.
- Symmetry and Spectroscopy an introduction to vibrational and electronic spectroscopy. Daniel C. Harris, etc.
- P. Bisson, G. Parodi, D. Rigos, J.E. Whitten, The Chemical Educator, 2006, Vol. 11, No. 2
- B. Schrader, Infrared and Raman Spectroscopy, VCH, 1995, ISBN:3-527-26446-9
- S.A. Borman, Analytical Chemistry, 1982, Vol. 54, No. 9, 1021A-1026A
- K. Nakamoto, Infrared Spectra of Inorganic and Coordination Compounds, 3rd edition. Wiley Intrsc John Wiley & Sons, New York London Sydney Toronto, 1978
- H.H. Claassen, C.L. Chernick, J.G. Malm, 1963 J. Am. Chem. Soc., 85, 1927
Problems
- What are the advantages and disadvantages for Raman spectroscopy, comparing with IR spectroscopy?
- Please briefly explain the mutual exclusive principle in Raman and IR spectroscopy.