
12.12: Normal Modes of Vibrations Describe how Molecules Vibrate
- Page ID
- 433954
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Normal modes of vibration
All molecules vibrate. The simplest vibration is the one that takes place between two atoms in a diatomic molecule. Vibrational motion in diatomic molecules is often discussed within the context of the simple harmonic oscillator in quantum mechanics. A diatomic molecule has only a single bond that can vibrate; we say it has a single vibrational mode. As you may expect, the vibrational motions of polyatomic molecules are much more complicated than those of a diatomic. First, there are more bonds that can vibrate; and secondly, in addition to stretching vibrations, the only type of vibration possible in a diatomic, we can also have bending and torsional vibrational modes. Since changing one bond length in a polyatomic will often affect the length of nearby bonds, we cannot consider the vibrational motion of each bond in isolation; instead we talk of normal modes of vibration involving the concerted motion of groups of bonds. As a simple example, the normal modes of a linear triatomic molecule are shown below.
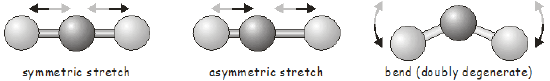
Once we know the symmetry of a molecule at its equilibrium structure, group theory allows us to predict the vibrational motions it will undergo using exactly the same tools we used above to investigate molecular orbitals. Each vibrational mode transforms as one of the irreducible representations of the molecule’s point group. Before moving on to an example, we will quickly review how to determine the number of vibrational modes in a molecule.
Molecular degrees of freedom – determining the number of normal vibrational modes
An atom can undergo only translational motion, and therefore has three degrees of freedom corresponding to motion along the \(x\), \(y\), and \(z\) Cartesian axes. Translational motion in any arbitrary direction can always be expressed in terms of components along these three axes. When atoms combine to form molecules, each atom still has three degrees of freedom, so the molecule as a whole has \(3N\) degrees of freedom, where \(N\) is the number of atoms in the molecule. However, the fact that each atom in a molecule is bonded to one or more neighboring atoms severely hinders its translational motion, and also ties its motion to that of the atoms to which it is attached. For these reasons, while it is entirely possible to describe molecular motions in terms of the translational motions of individual atoms (we will come back to this in the next section), we are often more interested in the motions of the molecule as a whole. These may be divided into three types: translational; rotational and vibrational.
Just as for an individual atom, the molecule as a whole has three degrees of translational freedom, leaving \(3N - 3\) degrees of freedom in rotation and vibration.
The number of rotational degrees of freedom depends on the structure of the molecule. In general, there are three possible rotational degrees of freedom, corresponding to rotation about the \(x\), \(y\), and \(z\) Cartesian axes. A non-linear polyatomic molecule does indeed have three rotational degrees of freedom, leaving \(3N - 6\) degrees of freedom in vibration (i.e \(3N - 6\) vibrational modes). In a linear molecule, the situation is a little different. It is generally accepted that to be classified as a true rotation, a motion must change the position of one or more of the atoms. If we define the \(z\) axis as the molecular axis, we see that spinning the molecule about the axis does not move any of the atoms from their original position, so this motion is not truly a rotation. Consequently, a linear molecule has only two degrees of rotational freedom, corresponding to rotations about the \(x\) and \(y\) axis. This type of molecule has \(3N - 5\) degrees of freedom left for vibration, or \(3N - 5\) vibrational modes.
In summary:
- A linear molecule has \(3N - 5\) vibrational modes
- A non-linear molecule has \(3N - 6\) vibrational modes.
Symmetry
Let’s work through an example: Ammonia (\(NH_3\)) with a \(C_{3v}\) symmetry. Consequently, all of the properties contained in the \(C_{3v}\) character table above are pertinent to the ammonia molecule.
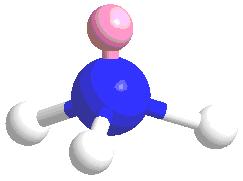
The principle axis is the axis that the highest order rotation can be preformed. In this case the z-axis pass through the lone pairs (pink sphere), which contains a \(C_3\) axis. The ?’s or mirror planes (\(\sigma_v\) parallel to z-axis & \(\sigma_h\) perpendicular to the z-axis). In ammonia there is no \(\sigma_h\) only three \(\sigma_v\)’s. The combination of \(C_3\) & \(\sigma_v\) leads to \(C_{3v}\) point group, which leads to the C3v character table.
The number of transitions is dictated by 3N-6 for non-linear molecules, so in the case of Ammonia, there will be \(3(4)-6=6\) vibrational modes. This can be confirmed by working through the vibrations of the molecule. This work is shown in the table below.
| \(C_{3v}\) | \(E\) | \(2C_3\) | \(3\sigma_v\) |
|---|---|---|---|
| \(\Gamma_{xyz}\) | 3 | 0 | 1 |
| Unmoved Atoms | 4 | 1 | 1 |
| \(\Gamma_\text{total}\) | 12 | 0 | 1 |
\(\Gamma_\text{total}\) is the total reducible form for the motions of ammonia. Using the character table for \(C_{3v}\), we can subtract the 3 translation motions defined by \(x\), \(y\), and \(z\), as well as the rotation motions defined by \(R_x\), \(R_y\), and \(R_z\):
| \(C_{3v}\) | \(E\) | \(2C_3\) | \(3\sigma_v\) |
|---|---|---|---|
| \(\Gamma_\text{total}\) | 12 | 0 | 1 |
| \(\Gamma_\text{translational}\) | 3 | 0 | 1 |
| \(\Gamma_\text{rotational}\) | 3 | 0 | -1 |
| \(\Gamma_\text{vibrational}\) | 6 | 0 | 1 |
| \( 2A_1 + 2E \) | |||
This gives us the reducible representation for the vibrational motion of ammonia (\(\Gamma_\text{vibrational}\)), which we can reduce down to there irreducible representations of \(2A_1\) and \(2E\). \(E\) is doubly degenerate, meaning two vibration modes each, totalling 6 vibrations. This calculation was done by using the character table to find out the rotation and translation values and what atoms move during each operation. Using the character table we can characterize the \(A_1\) vibration as IR active along the z-axis and raman active as well. The \(E\) vibration is IR active along both the x & y axis and is Raman active as well. From the character table the IR symmetries correspond to the x, y & z translations. Where the Raman active vibrations correspond to the symmetries of the d-orbitals.