
10.2: Physical and Spectroscopic Properties of Alkenes and Alkynes
- Page ID
- 22250

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Physical Properties
In general, the physical properties of alkenes are similar to those of alkanes. The data of Table 10-1 allow comparison of the boiling points, melting points, and densities of several alkenes with the corresponding alkanes that have the same carbon skeleton. Like the continuous-chain alkanes, the 1-alkenes form a homologous series of compounds that show regular changes in physical properties with increasing chain length. The boiling points, melting points, and densities of the simple alkynes (also included in Table 10-1) are somewhat higher than those of the corresponding alkanes or alkenes, and these properties also show regular changes as the chain length is increased.
Table 10-1: Comparison of Physical Properties of Alkanes, Alkenes, and Alkynes
Spectroscopic Properties of Alkenes
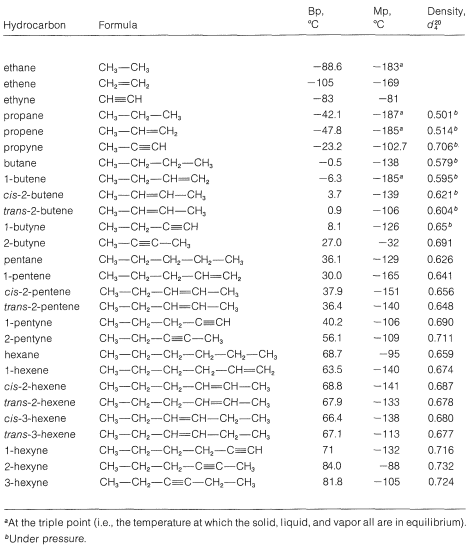
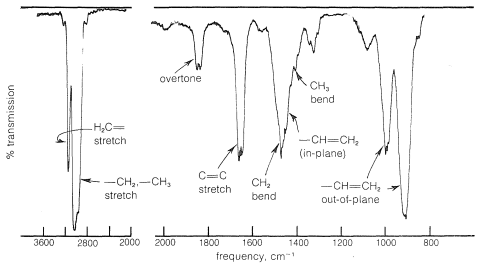
The infrared spectra of alkenes are sufficiently different from those of alkanes in most instances to make it possible to recognize when a double bond is present. For example, in the infrared spectrum of 1-butene (Figure 10-1) the absorption band near \(1650 \: \text{cm}^{-1}\) is characteristic of the stretching vibration of the double bond. In general, the intensity and position of this band depends on the structure of the alkene; it varies with the degree of branching at the double bond, with the presence of a second unsaturated group in conjugation with the first (i.e.,  or
or  ), and with the symmetry of the substitution of the double bond (see Section 9-7B). However, in many cases the absorption bands caused by the various modes of vibration of the alkenic \(\ce{C-H}\) bonds frequently are more useful for detecting a double bond and identifying its type than is the absorption band caused by \(\ce{C=C}\) stretch. With 1-butene, absorptions arising from the \(\ce{C-H}\) vibrations of the terminal \(\ce{=CH_2}\) group occur near \(3100 \: \text{cm}^{-1}\), \(1420 \: \text{cm}^{-1}\), and \(915 \: \text{cm}^{-1}\), and those of the \(\ce{-CH=}\) grouping near \(3020 \: \text{cm}^{-1}\), \(1420 \: \text{cm}^{-1}\), and \(1000 \: \text{cm}^{-1}\). In general, absorption bands at these frequencies are from the grouping \(\ce{-CH=CH_2}\). The bands near \(1420 \: \text{cm}^{-1}\) are due to in-plane bending, whereas those at \(915 \: \text{cm}^{-1}\) to \(1000 \: \text{cm}^{-1}\) arise from out-of-plane bending. The other intense absorptions, near \(1460 \: \text{cm}^{-1}\) and \(3000 \: \text{cm}^{-1}\), are due to \(\ce{C-H}\) vibrations of the \(\ce{CH_3CH_2}-\) group (see Section 9-7D). These illustrate a further point - namely, the positions of the infrared absorptions of alkyl \(\ce{C-H}\) bonds are significantly different from those of alkenic \(\ce{C-H}\) bonds.
), and with the symmetry of the substitution of the double bond (see Section 9-7B). However, in many cases the absorption bands caused by the various modes of vibration of the alkenic \(\ce{C-H}\) bonds frequently are more useful for detecting a double bond and identifying its type than is the absorption band caused by \(\ce{C=C}\) stretch. With 1-butene, absorptions arising from the \(\ce{C-H}\) vibrations of the terminal \(\ce{=CH_2}\) group occur near \(3100 \: \text{cm}^{-1}\), \(1420 \: \text{cm}^{-1}\), and \(915 \: \text{cm}^{-1}\), and those of the \(\ce{-CH=}\) grouping near \(3020 \: \text{cm}^{-1}\), \(1420 \: \text{cm}^{-1}\), and \(1000 \: \text{cm}^{-1}\). In general, absorption bands at these frequencies are from the grouping \(\ce{-CH=CH_2}\). The bands near \(1420 \: \text{cm}^{-1}\) are due to in-plane bending, whereas those at \(915 \: \text{cm}^{-1}\) to \(1000 \: \text{cm}^{-1}\) arise from out-of-plane bending. The other intense absorptions, near \(1460 \: \text{cm}^{-1}\) and \(3000 \: \text{cm}^{-1}\), are due to \(\ce{C-H}\) vibrations of the \(\ce{CH_3CH_2}-\) group (see Section 9-7D). These illustrate a further point - namely, the positions of the infrared absorptions of alkyl \(\ce{C-H}\) bonds are significantly different from those of alkenic \(\ce{C-H}\) bonds.
The double bonds of an alkene with no alkenic hydrogens are difficult to detect by infrared spectroscopy and in such cases Raman spectroscopy is helpful (see Section 9-8).
The infrared absorption of 1-butene that occurs at \(1830 \: \text{cm}^{-1}\) (Figure 10-1) falls in the region where stretching vibrations of alkene bonds usually are not observed. However, this band actually arises from an overtone (harmonic) of the \(\ce{=CH_2}\) out-of-plane bending at \(915 \: \text{cm}^{-1}\). Such overtone absorptions come at exactly twice the frequency of the fundamental frequency, and whenever an absorption like this is observed that does not seem to fit with the normal fundamental vibrations, the possibility of its being an overtone should be checked.
With regard to electronic spectra, a \(\pi\) electron of a simple alkene can be excited to a higher energy \(\left( \pi^* \right)\) state by light of wavelength \(180 \: \text{nm}\) to \(100 \: \text{nm}\). However, many other substances absorb in this region of the spectrum, including air, the quartz sample cell, and most solvents that might be used to dissolve the sample, and as a result the spectra of simple alkenes are not obtained easily with the usual ultraviolet spectrometers. When the double bond is conjugated as in 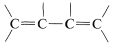 or
or 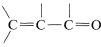 , then the wavelengths of maximum absorption shift to longer wavelengths and such absorptions are determined more easily and accurately (also see Section 9-9B).
, then the wavelengths of maximum absorption shift to longer wavelengths and such absorptions are determined more easily and accurately (also see Section 9-9B).
In proton nmr spectra, the chemical shifts of alkenic hydrogens are toward lower fields than those of alkane hydrogens and normally fall in the range of \(4.6\)-\(5.3 \: \text{ppm}\) relative to TMS (see section 9-10E and Table 9-4). Spin-spin couplings of alkenic hydrogens are discussed in Section 9-10G and 9-10J.
Spectroscopic Properties of Alkynes
The infrared spectrum of a monosubstituted alkyne such as ethynylbenzene, \(\ce{C_6H_5C \equiv CH}\) (Figure 10-4), has a strong band near \(3300 \: \text{cm}^{-1}\), which is characteristic of the carbon-hydrogen stretching vibration in the grouping \(\ce{\equiv C-H}\). At a lower frequency (longer wavelength) around \(2100 \: \text{cm}^{-1}\), there is a band associated with the stretching vibration of the triple bond (also see Figure 9-36). Therefore the presence of the grouping \(\ce{-C \equiv CH}\) in a molecule may be detected readily by infrared spectroscopy. However, the triple bond of a disubstituted alkyne, \(\ce{R-C \equiv C-R}\), is detected less easily because there is no \(\ce{\equiv C-H}\) absorption near \(3300 \: \text{cm}^{-1}\), and furthermore the \(\ce{C \equiv C}\) absorption sometimes is of such low intensity that it may be indiscernible. Raman spectroscopy (Section 9-8) or chemical methods must then be used to confirm the presence of a triple bond.
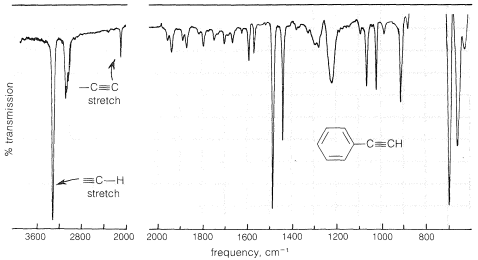
Alkynes, like alkenes, undergo electronic absorption strongly only at wavelengths in the relatively inaccessible region below \(200 \: \text{nm}\). However, when a triple bond is conjugated with one or more unsaturated groups, radiation of longer wavelength is absorbed. To illustrate, ethyne absorbs at \(150 \: \text{nm}\) and \(173 \: \text{nm}\), whereas 1-buten-3-yne \(\left( \ce{CH_2=CH-C \equiv CH} \right)\) absorbs at \(219 \: \text{nm}\) and \(227.5 \: \text{nm}\). The effects of such conjugation on spectra is discussed in more detail in Section 9-9B.
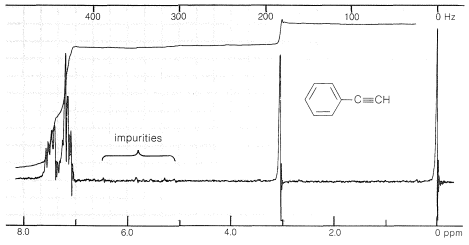
The proton nuclear magnetic resonance spectrum of ethynylbenzene is shown in Figure 10-5. The peaks near \(435 \: \text{Hz}\) and \(185 \: \text{Hz}\) correspond to resonances of the phenyl and \(\ce{\equiv C-H}\) protons, respectively. The difference in chemical shift between the two types of protons is considerably larger than between alkenic and aromatic protons come into resonance at higher magnetic fields (i.e., they are subject to more diamagnetic shielding, Section 9-10E) than alkenic or aromatic protons. In fact, the \(\ce{\equiv C-H}\) protons of alkynes have chemical shifts approaching those of alkyl protons. (Also see Figure 9-36.)
The mass spectra of alkenes and alkynes usually give distinct molecular ions; however, the fragmentation is often complex and not easily interpreted.
References
John D. Robert and Marjorie C. Caserio (1977) Basic Principles of Organic Chemistry, second edition. W. A. Benjamin, Inc. , Menlo Park, CA. ISBN 0-8053-8329-8. This content is copyrighted under the following conditions, "You are granted permission for individual, educational, research and non-commercial reproduction, distribution, display and performance of this work in any format."