
4.4: Nucleophilic substitution and elimination reactions
- Page ID
- 416447

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)- Identify nucleophiles and electrophiles, their similarities, and differences from acids and bases.
- Understand
- Learn some examples of
Nucleophile and electrophile
Nucleophile is a neutral or anionic specie that can donate a lone pair or \(\pi\) bonding electrons to make a covalent bond.
Examples of nucleophile are negative or partial negative (\(\delta{-}\)) atoms with lone pair or \(\pi\) bond in the following \(\ce{H3{N}{_{\bullet}^{\bullet}}}\), \(\ce{H2\overset{\!\bullet\bullet}{O}{_{\bullet}^{\bullet}}}\), \(\ce{H\overset{\!\!\!\bullet\bullet}{\underset{\bullet\bullet}{O}{_{\bullet}^{\bullet}}}^{-}}\), and \(\ce{H2C=CH2}\).
Electrophile is an electron-deficient atom of a neutral or cationic species that can receive an electron pair to make a covalent bond.
Examples of electrophile are positive charge or partial positive (\(\delta{+}\)) charge \(\ce{C's}\) or \(\ce{H's}\) in the following species: \(\ce{(CH3)3\overset{+}{C}}\), \(\ce{\overset{\delta{+}}{C}H3{-}\overset{\delta{-}}{Cl}}\), \(\ce{(CH3)2\overset{\delta{+}}{C}{=}\overset{\delta{-}}{O}}\), and \(\ce{\overset{\delta{+}}{H}{-}\overset{\delta{-}}{Cl}}\).
Differences between nucleophile-electrophile and acid-base
The base is a substance that donates a pair of electrons to a proton to make a covalent bond. So, a base is a sub-class of nucleophiles and an acidic proton is a subclass of electrophiles. Acid-base reactions are fast reactions. In acid-base reactions, emphasis is on thermodynamic, specifically on the equilibrium constant K quantified in terms of pKa, i.e., smaller the pKa means larger K and stronger acid. Nucleophiles donate electrons to an electron-deficient carbon or some atom usually other than a proton. Emphasis in nucleophilic-electrophilic reaction is on the kinetics, a good nucleophile reacts faster and a poor nucleophile reacts slower.
Similarities and differences in nucleophilicity and basicity
The similarity is that the stronger bases are stronger nucleophiles when compared within the same row of the periodic table. For example, basicity follows the order \(\overrightarrow{\ce{HO^{-}<NH2^{-}<F^{-}}}\) and their nucleophilicity follows the same order. Similarly, anionic species, e.g., \(\ce{NH2^{-}}\), \(\ce{HO^{-}}\) and \(\ce{RO^{-}}\) are stronger bases and good nucleophiles compared to the corresponding neutral species of the same element, i.e., \(\ce{NH3}\), \(\ce{H2O}\) and \(\ce{ROH}\).
The difference is that the basicity and nucleophilicity are affected differently when comparing atoms of different rows. For example, hydride ion (\(\ce{H^{-}}\)) from the 1st row is a stronger base but a poor nucleophile. the opposite is true for the 3rd-row and higher elements, e.g., \(\ce{SH^{-}}\), \(\ce{Cl^{-}}\), \(\ce{Br^{-}}\). and \(\ce{I^{-}}\) are week bases but usually good nucleophiles. Unlike basicity, nucleophilicity is affected by steric factors and solvents. Most of these differences are related to the fact that in acid-base reactions, the electron recipient is the 1s orbital of a proton, and in nucleophilic-electophilic reactions, the electron recipient is 2s, 2p, or larger orbital of carbon or some other element. The details of these factors are out of the scope of this book. Base strength is described as stronger or weak, and nucleophilic strength is described as good or poor.
Nucleophilic substitution mechanisms
In a nucleophilic substitution reaction, a nucleophile ( \(\ce{Nu{_{\bullet}^{\bullet}}^{-}}\) ) attacks and makes a covalent bond with \(\delta{+}\) atom of the target molecule, called substrate (\(\ce{\overset{\delta{+}}{R-}\overset{\delta{-}}{X}}\)). The polar bond of the target molecule breaks heterolytically, leaving the bonding electrons with the more electronegative end called leaving group (\(\ce{X{_{\bullet}^{\bullet}}^{-}}\)), as shown in the following generalized reaction.
\[\ce{{Nu}{_{\bullet}^{\bullet}}^{-} + \overset{\delta{+}}{R-}\overset{\delta{-}}{X} -> {\overset{\delta{+}}{R-}\overset{\delta{-}}{Nu}} + X{_{\bullet}^{\bullet}}^{-} }\nonumber \]
Since one nucleophile, i.e., \(\ce{Nu{_{\bullet}^{\bullet}}^{-}}\), replaces another nucleophile, i.e., \(\ce{X{_{\bullet}^{\bullet}}^{-}}\) on the substrate, it is called a nucleophilic substitution reaction. The nucleophilic substitution reactions are based on the fact the incoming nucleophile has a stronger tendency to make a covalent bond with the electrophilic center than the leaving group. In other words, the incoming nucleophile is stronger and the leaving group is a weaker nucleophile. Mechanisms of the nucleophilic substitution reactions are described below.
Nucleophilic substitution bimolecular (SN2)
One mechanism for nucleophilic substitution reaction is concerted bond-making and breaking in a single step, as shown below.
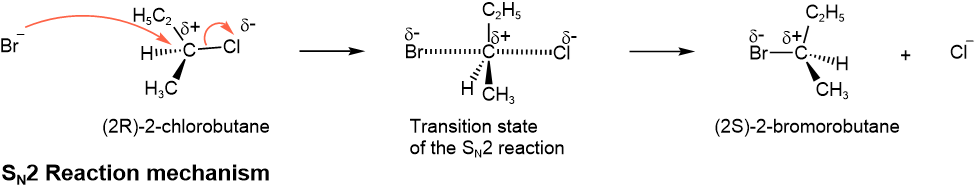
The incoming nucleophile approaches the electrophilic \(\ce{\overset{\delta{+}}{C}}\) from the side opposite to the leaving group. The incoming nucleophile starts making the bond and the leaving group starts breaking the bond simultaneously. The other three groups pointing away from the leaving group, start moving to the other side, away from the incoming nucleophile. In the transition state, the three groups acquire a trigonal planar arrangement perpendicular to the bond being broken and formed. This reaction mechanism is called SN2, where S is for substitution, N is for nucleophilic, and 2 is for bimolecular.
- If the electrophilic \(\ce{\overset{\delta{+}}{C}}\) is a chiral center, its configuration is inverted in the product of SN2 reaction.
- This elementary step of SN2 reaction involves two reactants in its rate-determining step, the incoming nucleophile, and the substrate, and it is a bimolecular reaction.
The rate of an SN2 reaction is directly proportional to the substrate concentration and the nucleophile concentration. The rate depends on the substrate's structure, the nature of the leaving group, the nature of the nucleophile, and the solvent, as described next.
Effect of the structure of the substrate
The rate follows the following order with respect to the nature of the electrophilic \(\ce{\overset{\delta{+}}{C}}\): \(\overleftarrow{\text{ methyl > primary > secondary > tertiary}}\). The steric hindrance posted by the substrate to the nucleophile explains it, which follows the same order, as illustrated below.
| Type of electrophile carbon | Methyl | Primary | Secondary | Tertiary |
| Model |  |
 |
 |
 |
| Structures with mechanism arrows |  |
 |
 |
 |
| The relative SN2 reaction rate | 145 | 1 | 0.008 | no reaction |
Effect of the nucleophile
Good nucleophiles react faster. Particularly, anionic nucleophiles, like \(\ce{HO^{-}}\) or \(\ce{RO^{-}}\) are employed for SN2 as they react much faster than their corresponding neutral counterparts, i.e., \(\ce{H2O}\) or \(\ce{ROH}\), as shown in the following example.

Effect of the leaving group
Leaving group propensity has a trend opposite to the basicity of the leaving group, i.e., good leaving groups are weaker bases. For example, the basicity of halide ions follows this order: \(\overleftarrow{\ce{F^{-} > Cl^{-} > Br^{-} > I^{-}}}\) but the rate of reaction of alkyl halides, under the same conditions, follows the opposite trend, i.e., \(\overrightarrow{\ce{R-F < R-Cl < R-Br < R-I}}\). The effect of the leaving group on the rate of the reaction is shown in the following table.
| Reaction | Relative rate |
| \(\ce{HO^{-} + RCH2-F -> RCH2-OH + F^{-}}\) | 1 |
| \(\ce{HO^{-} + RCH2-Cl -> RCH2-OH + Cl^{-}}\) | 200 |
| \(\ce{HO^{-} + RCH2-Br -> RCH2-OH + Br^{-}}\) | 10,000 |
| \(\ce{HO^{-} + RCH2-I -> RCH2-OH + I^{-}}\) | 30,000 |
Effect of the solvent
The solvent is needed to dissolve both the substrate and the nucleophile. The substrate is a polar compound like \(\ce{\overset{\delta{+}}{CH3-}\overset{\delta{-}}{Br}}\) and the nucleophile is usually in the form of an ionic solid like \(\ce{Na^{+}OH^{-}}\) or \(\ce{CH3O^{-}Na^{+}}\). Nonpolar solvents do not work because polar and ionic substances do not dissolve in nonpolar solvents. Polar solvents can dissolve polar and ionic compounds. Polar solvents fall into two categories:
- polar protic solvent that have an acidic proton like water (\(\ce{H2O}\)) and methanol (\(\ce{CH3-OH}\)), and
- polar aprotic like acetone ( \(\ce{(CH3)2\overset{\delta{+}}{C}{=}\overset{\delta{-}}{O}}\) ) and dimethylsulfoxide ( \(\ce{(CH3)2\overset{\delta{+}}{S}{=}\overset{\delta{-}}{O}}\) }\)) which do not have an acidic proton. Their \(\delta{+}\) end is at the center of the molecule, surrounded by bulky groups.
Polar protic solvent dissolves ionic compounds by ion-dipole interactions forming a layer of solvent around cation and anion. They are not suitable for SN2 reactionas the solvent layer prevents nucleophiles from approaching electrophilic \(\ce{\overset{\delta{+}}{C}}\).
Aprotic solvents dissolve the ionic compound by the ion-dipole interaction but leave the anion, i.e., nucleophile, almost free. This is because the anion is prevented from approaching the \(\delta{+}\) pole of the solvent due to steric hindrance, as illustrated in Figure \(\PageIndex{1}\). Recall that electrostatic force is inversely proportional to the square of the distance between the +ve charge and the -ve charge, i.e., the longer the distance, the weaker the force.
Polar aprotic solvents are suitable for SN2 reaction as the nucleophiles are relatively free to approach electrophilic \(\ce{\overset{\delta{+}}{C}}\) of the substrate.

Examples of SN2 reactions
Alkyl halides are derived from alkanes by free radical reactions. The alkyl halides are then converted to a number of other classes of organic compounds by SN2 reaction, e.g., ethyl bromide can be converted to:
alcohol: \(\ce{CH3CH2Br + OH^{-} -> CH3CH2OH + Br^{-}}\),
a thiol: \(\ce{CH3CH2Br + HS^{-} -> CH3CH2SH + Br^{-}}\),
an ether: \(\ce{CH3CH2Br + CH3CH2O^{-} -> CH3CH2OCH2CH3 + Br^{-}}\),
a thioether: \(\ce{CH3CH2Br + CH3CH2S^{-} -> CH3CH2SCH2CH3 + Br^{-}}\),
an amine: \(\ce{CH3CH2Br + ^{-}NH2 -> CH3CH2NH2 + Br^{-}}\),
a nitrile: \(\ce{CH3CH2Br + ^{-}C≡N -> CH3CH2C≡N + Br^{-}}\), and
and an alkyne: \(\ce{CH3CH2Br + ^{-}C≡CCH3 -> CH3CH2C≡CCH3 + Br^{-}}\).
Elimination bimolecular (E2)
Recall that the \(\ce{C}\) connected to a functional group is \(\alpha\ce{C}\), and the one adjacent to it is \(\beta\ce{C}\). Also, recall that \(\ce{\overset{\delta{+}}{H-}\overset{\delta{-}}{Br}}\) is a strong acid because the acidic polar leaves electron on elecronegative \(\ce{Br}\). Similarly, protons on \(\beta\ce{C}\) are acidic because they can send the bonding electrons to \(\ce{Br}\) through the following mechanism.

Nucleophiles are bases at the same time. The base \(\ce{HO^{-}}\) attacks \(\beta\ce{H}\), the \(\ce{H-C}\) bonding electrons establish a \(\pi\)-bond between \(\alpha\ce{C}\) and \(\beta\ce{C}\), and the \(\alpha\ce{C}\) let go the leaving group \(\ce{Br^{-}}\). Since a \(\beta\ce{H}\) is eliminated along with the leaving group, This reaction mechanism is called \(\beta\)-elimination. It is a bimolecular reaction because of two species involved in this elementary step: the base and the substrate. This specific \(\beta\)-elimination is called elimination bimolecular (E2), where E stands for elimination and 2 for bimolecular.
SN2 vs. E2
SN2 and E2 reactions compete because the same reagent is nucleophile in SN2 and base in the E2 reaction. However, the effect of the structure of the substrate is opposite in SN2 and E2. The structure of substrate affects SN2 in this order: \(\overleftarrow{\text{ methyl > primary > secondary > tertiary}}\). Tertiary substrates do not react by SN2 mechanics because the nucleophile is prevented from approaching \(\alpha\ce{C}\) by steric hindrance. However, \(\beta\ce{H}\) is still exposed and easily approachable by the base, as shown below.

Further, E2 is facilitated by lowering steric crowding around the \(\alpha\ce{C}\). Therefore, tertiary substrates yield E2 product under the SN2 conditions. The steric crowding around \(\alpha\ce{C}\) of secondary substrates is less than tertiary. So, E2 and SN2 compete, and elimination and substation products are formed from the secondary substrates. There is no steric crowding in the case of primary or methyl substrates. So, SN2 dominated over E2, and substitution product is formed almost exclusively in the case of primary and methyl substrates. The effect of the structure of the substrate on E2 reaction is in this order: \(\overrightarrow{\text{ methyl < primary < secondary < tertiary}}\), which is the opposite of SN2. E2 reactions synthesize alkenes from alkyl halides, as in the above example, and form similar functional groups that will be described later.
Substitution nucleophilic unimolecular SN1
Ionic compounds like \(\ce{Na^{+}Cl^{-}}\) dissociate into ions in polar protic solvents like \(\ce{H2O}\) better than in polar aprotic solvents like. This is because unlike polar aprotic solvents solvating only cations, polar protic solvents solvent both cations and anions. Polar covalent bonded compounds like \(\ce{\overset{\delta{+}}{H-}\overset{\delta{-}}{Br}}\) also dissociate in polar portic solvent by the same mechanism. Polar organic compounds like \(\ce{\overset{\delta{+}}{H3C-}\overset{\delta{-}}{Br}}\) have less tendency to dissociate. However, if the substrate is tertiary like \(\ce{(H3C)3\overset{\!\!\!\!\delta{+}}{C-}\overset{\delta{-}}{Br}}\), the effect of steric crowding on \(\alpha\ce{C}\) and the dissociation ability of the polar protic solvent together are strong enough to dissociate polar organic compounds, as in 1st step of the mechanism shown in Figure \(\PageIndex{2}\).

The carbonation produced in the 1st step, e.g., \(\ce{(H3C)3C^{+}}\) in this case, is a strong electrophile and reacts with any nucleophile around, including solvent molecules, e.g., (\(\ce{H2O}\) in this case. Since the 2nd step in non-selective, the solvent wins over any other nucleophile in the system because of its higher concentration. The neutral nucleophile becomes a cationic group in the product of the 2nd step, e.g., \(\ce{R-OH2^{+}}\) in this case. The strong acid like \(\ce{R-OH2^{+}}\) donates its proton to any base (\(\ce{{_{\bullet}^{\bullet}}B}\)) in the medium, including the solvent, e.g., \(\ce{H2O}\) in this case.
The overall reaction is substitution nucleophilic. The overall reaction is unimolecular proportional to the substrate concentration in the 1st step. So, this mechanism is called substitution nucleophilic unimolecular SN1, where S stands for substitution, N or nucleophilic, and 1 for unimolecular.
- SN1 is unimolecular.
- The carbonation intermediate is trigonal planar and the nucleophile can attack it from either side to make a new covalent bond. Therefore, if SN1 reaction happens on a chiral carbon, around 50% of the product retains the configuration of the reactant, and the other 50% has the inverted configuration, i.e., the product is a racemic mixture.
- SN1 Reaction takes place on tertiary alkyl halides. Secondary and primary alkyl halides do not undergo SN1 reactions.
Elimination unimolecular (E1)
The carbocation formed in SN1 reaction has acidic protons on \(\beta\ce{C}\) because the electrons left on the conjugate base are stabilized as a \(\pi\)-bond, as in the mechanism shown below.

This mechanism eliminates a leaving group and a proton \(\beta\) to the leaving group. The 1st step, which is the rate-determining step, is unimolecular. This mechanism is called elimination unimolecular (E1), where E stands for elimination and 1 for unimolecular. E1 always competes with SN1 because they have a common first step.
What decides the reaction will happen by
All of the The solvent and the nucleophile determine SN2 or SN1 condition.
Strong nucleophiles, usually anionic like \(\ce{HO^{-}}\), \(\ce{RO^{-}}\), and polar aprotic solvent like acetone (\(\ce{(CH3)2C=O}\)) or dimethylsulfoxide (\(\ce{(CH3)2S=O}\)) define SN2 and E2 condition.
The substrate dictates SN2, E2, or both will happen: SN2 happens on the primary or methyl substrate, E2 happens on the tertiary substrate, and both happen simultaneously on a secondary substrate. SN2 and E2 reactions do not take place in polar protic solvents.
Polar protic solvents like water (\(\ce{H2O}\)) or methanol (\(\ce{CH3-OH}\)) and neutral nucleophiles like \(\ce{H2O}\), \(\ce{ROH}\) define SN1 and E1 condition.
Anionic nucleophiles can not exist in protic sovlents due to acid-base neutralization reactions between them. Neutral species are poor nucleophiles but are OK in SN1 or E1 reactions because they are not involved in the rate-determining step. The following data on the dissociation rate of tertiary butyl chloride shows the effect of polar protic solvent on the rate-determining step of SN1 and E1 reactions.

| Solvent | Polarity (Dielectric constant) | Protic or aprotic | Relative rate |
|---|---|---|---|
| Water (\(\ce{H2O}\)) | 78 | Polar protic | 40 |
| Ethanol (\(\ce{CH3CH2-OH}\)) | 24 | Polar protic | 1 |
| Acetone (\(\ce{(CH3)2C=O}\) | 21 | Polar aprotic | 0.005 |
SN1 and E1 usually compete as both have the same rate-determining step. Tertiary substrate easily react by SN1 and E1 mechanism. Secondary substrate may or may not take place by SN1 and E1. For example, secondary alkyl halides do not react, but secondary alcohols and ethers react by SN1 and E1. Primary substrates usually do not react by SN1 and E1 mechanisms.
Effect of leaving group on
Recall that the stronger the acid, the weaker the conjugate base, and voice versa. For example, \(\ce{HI}\) is a strong acid and \(\ce{I^{-}}\) is a weak base, while \(\ce{HF}\) is a weak acid and \(\ce{F^{-}}\) is a strong base. In other words, strong bases do not tend to protons, and weak bases easily leave protons in acid-base reactions. The same applies to leaving groups in reactions.
Strong bases are poor leaving groups, and weak bases are good leaving groups in reactions.
The basicity and hence the leaving propensity of leaving groups follows these trends:
- Basicity decreases from top to bottom in a group of periodic table. For example, the base strength of halides follows this order: \(\overleftarrow{\ce{F^{-} > Cl^{-} > Br^{-} > I^{-}}}\). Leaving propensity follows the opposite trend i.e., \(\ce{-I}\) is the best-leaving group, while \(\ce{-F}\) is the worst leaving group. \(\ce{-F}\) does not act as a leaving group.
- Basicity decreases from right to left in a row of the periodic table. For example, the base strength of 2nd row elements follows this order: \(\overleftarrow{\ce{F^{-} < OH^{-} < NH2^{-} < CH3^{-}}}\). Leaving propensity follows the opposite trend, i.e., \(\ce{-OH}\) \(\ce{-NH2}\). \(\ce{-CH3}\) are the worst leaving group than \(\ce{-F}\) and do not act as leaving group.
- Basicity decreases upon protonation in the cases of amphoteric species. For example, \(\ce{H2O}\) is a weaker base than \(\ce{OH^{-}}\). That is why, \(\ce{-OH}\) is not a leaving group, but \(\ce{-\overset{+}{O}H2}\) is a good leaving group that leaves as a weak base \(\ce{H2O}\).
Nucleophilic substitution and elimination reactions of alcohols, ethers, amines, and sulfur compounds
Reactions of alcohols
Substitution reactions
The \(\ce{O}\) of an alcohol (\(\ce{R-OH}\)) is protonated by a strong acid to convert it from not leaving group ((\(\ce{-OH}\)) to a good leaving group ((\(\ce{-\overset{+}{O}H2}\)). The protonated alcohols under go SN1 or E1 reactions, except when the substrate is primary. that undergoes SN2 or E2 reactions. For example, primary, secondary, and tertiary alcohols undergo nucleophilic substitution with \(\ce{HCl}\), \(\ce{HBr}\), or \(\ce{HI}\) as shown below.
\(\ce{(CH3)3C-OH + HBr <=> (CH3)3C-{\overset{+}{O}}H2 + Br^{-} ->[\Delta] (CH3)3C-Br + H2O}\nonumber\) by SN1 mechanism
\(\ce{(CH3)2CH-OH + HBr <=> (CH3)2CH-{\overset{+}{O}}H2 + Br^{-} ->[\Delta] (CH3)2CH-Br + H2O}\nonumber\) by SN1 mechanism
\(\ce{CH3CH2-OH + HBr <=> CH3CH2-{\overset{+}{O}}H2 + Br^{-} ->[\Delta] CH3CH2-Br + H2O}\nonumber\) by SN2 mechanism
Elimination reactions may compete with substitution reactions described above, but the alkene products of elimination add \(\ce{HCl}\), \(\ce{HBr}\), or \(\ce{HI}\) and end in the same final product as the substitution products. These reactions of alkens will be described in a later section.
Elimination reactions
Inorder to perform elimination reactions, the \(\ce{O}\) of alcohols (\(\ce{R-OH}\)) is protonated by sulfuric acid (\(\ce{H2SO4}\)) in water solution. The conjugate base of sulfuric, i.e., \(\ce{[HSO4]^{-}}\) is a weak nucleophile and does not cause the substitution reaction. Water is a nucleophile, but its creation does not change the intermediate. However, water acts as a base picking up \(\beta\)\(\ce{H}\), causing elimination reactions, as shown below.
\(\ce{(CH3)3C-OH <=>[H2SO4 + H2O] (CH3)3C-{\overset{+}{O}}H2 ->[\Delta +HSO4^{-} + H2O] (CH3)2C=CH2 + H2O}\nonumber\) by E1 mechanism
\(\ce{(CH3)2CH-OH <=>[H2SO4 + H2O] (CH3)2CH-{\overset{+}{O}}H2 ->[\Delta +HSO4^{-} + H2O] CH3CH=CH2 + H2O}\nonumber\) by E1 mechanism
\(\ce{CH3CH2-OH <=>[H2SO4 + H2O] CH3CH2-{\overset{+}{O}}H2 ->[\Delta +HSO4^{-} + H2O] CH2=CH2 + H2O}\nonumber\) by E2 mechanism
These reactions are called dehydration of alcohols that convert alcohols to alkenes.
Alcohols are common intermediates in biochemical reactions. However, unlike in a chemical laboratory, strong acids like \(\ce{HBr}) or \(\ce(H2SO4) needed to activate alcohol groups do not survive under physiological conditions. Phosphoric acid and its anhydrides, i.e., pyrophosphoric acid and triphosphoric acids are weak acids and their conjugate bases, i.e., phosphate, pyrophosphate, and triphosphate, are weak bases and good leaving groups. Therefore, biological systems convert alcohols into phosphate or pyrophaste esters by reacting with adenosine triphosphate (ATP). It allows them to participate in the nucleophilic substitution reactions under physiological conditions as shown below.

Reactions of ethers
Like alcohols, the \(\ce{O}\) of ethers (\(\ce{R-O-R'}\)) is protonated by a strong acid to convert it from not leaving group ((\(\ce{-OR'}\)) to a good leaving group ((\(\ce{-\overset{+}{O}HR}\)). For example, ethers undergo nucleophilic substitution reactions with \(\ce{HCl}\), \(\ce{HBr}\), or \(\ce{HI}\) producing alcohols and an alkyl halides, as shown below.
\(\ce{(CH3)3C-O-C(CH3)3 + HBr <=> (CH3)3C-{\overset{+}{O}}HC(CH3)3 + Br^{-} ->[\Delta] (CH3)3C-Br + (CH3)3C-OH}\nonumber\) by SN1 mechanism
\(\ce{CH3CH2-O-CH3 + HBr <=> CH3CH2-{\overset{+}{O}}HCH3 + Br^{-} ->[\Delta] CH3CH2-OH + CH3-Br}\nonumber\) by SN2 mechanism
Ethylene oxide is a three-membered cyclic ether. It is a colorless gas with a boiling point of 11 oC. The \(\ce{C-O}\) bonds in ethylene oxide are unstable due to angle strain. Therefore, the ether groups in ethylene oxide act as excellent leaving groups because angle strain is released. Ethylene oxide reacts fast by SN2 mechanism with amino (\(\ce{-NH2}\)) and sulfhydryl (\(\ce{-SH}\)) groups that are commonly present in biochemicals, as shown below.
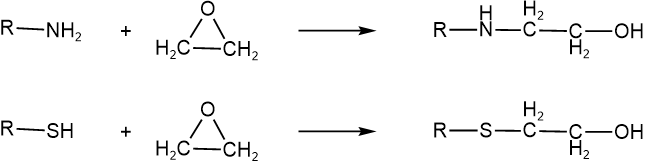
These reactions modify the biochemicals, leading to the death of microorganisms. Ethylene oxide is used as a fumigant in foods and textiles and to sterilize surgical instruments in hospitals.
Reactions of amines
Amine \(\ce{-NH2}\) and also its protonated form \(\ce{-NH3^{+}}\) are poor as leaving groups and do not act as leaving groups. However, amines are good nucleophiles and act as incoming nucleophiles in various reactions, e.g., in SN2 reactions with alkyl halides, as shown below.
\(\ce{CH3CH2-Br + CH3-NH2 -> CH3CH2-{\overset{+}{N}}H2-CH2CH3 + Br^{-}}\) an SN2 reaction
Reactions of thiols
Thiolate ion is a good nucleophile for SN2 reactions as shown in the following example.
\(\ce{CH3-\overset{\bullet\bullet}{\underset{\bullet\bullet}{S}}^{-} + CH3-I -> CH3-{\overset{\bullet\bullet}{S}}-CH3 + I^{-}}\) an SN2 reaction
Methyl (\(\ce{\overset{\delta{+}}{C}H3{-}\overset{\delta{-}}{X}}\) is the best electrophilic site, and iodide (\(\ce{-I}\)) is the best-leaving group that makes methyliodide (\(\ce{\overset{\delta{+}}{C}H3{-}\overset{\delta{-}}{I}}\)) the best substrate for methylating any nucleophile in an SN2 reactions, as shown in the following examples.
\(\ce{CH3-\overset{\bullet\bullet}{\underset{\bullet\bullet}{S}{_{\bullet}^{\bullet}}}^{-} + CH3-I -> CH3-\overset{\bullet\bullet}{\underset{\bullet\bullet}{S}}-CH3 + I^{-}}\) an SN2 reaction
Sulfur is a larger atom that can accommodate three alkyl groups. Therefore, dimethyl sulfide produced in the above reaction can be methylated one more time, producing a solfonium salt, as shown below.
\(\ce{CH3-\overset{\bullet\bullet}{\underset{\bullet\bullet}{S}}-CH3 + CH3-I -> (CH3)3{\overset{\bullet\bullet}{S}}^{+} I^{-}}\) an SN2 reaction
Dimethyl sulfide (\(\ce{(CH3)2{\overset{\bullet\bullet}{S}}^{+}{-}}\)) is an excellent leaving group that makes trimethylsulfonium ion (\(\ce{(CH3)3{\overset{\bullet\bullet}{S}}^{+}}\)) an excellent methylating agent, as shown in an example below.
\(\ce{HO^{-} + (CH3)3{\overset{\bullet\bullet}{S}}^{+} -> CH3-OH + CH3-\overset{\bullet\bullet}{\underset{\bullet\bullet}{S}}-CH3}\) an SN2 reaction
methyliodide (\(\ce{H3C-I}\)) is a common methylating agent in laboratory,- but it is not available in biological systems. S-adenosylmethionine (SAM), which is similar to trimethylsulfonium ion (\(\ce{(CH3)3{\overset{\bullet\bullet}{S}}^{+}}\)) is a common methylating agent in biological systems, as shown in an example below.

In the above example of a biochemical reaction, noradrenaline hormone is converted into a more potent adrenaline hormone by methylation reaction using SAM as a methylating agent.