
Metalloproteases
- Page ID
- 2359
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Metalloproteases (metallo, metal) are members of a clan of proteases that contain a metal ion at their active site which acts as a catalyst in the hydrolysis peptide binds.1 The metallic core of each enzyme is the location of the specific reaction performed by the enzyme, in the case of metalloproteases, the cleavage of peptide bonds within proteins. The most common metal ion metalloproteases utilize is a zinc ion (Zn2+).2 Other transition metals have been found at active sites, such as Co2+ and Mn2+, and some have been used to restore function in zinc-metalloproteases in which the Zn2+ core has been removed.3 Generally, metal ions are bound in a nearly tetrahedral conformation at the active site. Three amino acid ligands, usually charged residues, associate with the metal core along with one water molecule which is used for hydrolysis.4
There are two major divisions of metalloproteases: metalloendopeptidases and metalloexopeptidases. Each division is named for the region of the hydrolysis-targeted protein at which the reaction takes place.5,6 Within these divisions are nested more highly specified target sites and conserved catalytic residues wholly dependent on the nature of the enzyme.1,2,7
Metalloendopeptidase: Thermolysin
Background
Thermolysin (TLN; EC 3.4.24.27, Figure 1) is a 34.6 kDa Zn2+-endopeptidase secreted by the bacterium Bacillus thermoproteolyticus.8,9 TLN and TLN-like proteins are used by bacteria to break down exogenous proteins for nutrition and as virulence factors aiding in host colonization and tissue degradation.10,11 TLN is active in the hydrolysis of internal peptide bonds on the N-terminal side of large hydrophobic amino acids, between leucine, isoleucine, or phenylalanine. Thermolysin was the first metalloproteases to be completely sequenced.12 Commercially, TLN is used as a nonspecific protease (within its cleavage site specificity) in peptide sequencing and is used in the production of the artificial sweetener aspartame.13

Figure 1. General structure of thermolysin (PDBID 3DNZ). Alpha-helices in blue, beta-sheets in red, Zn2+ ion in yellow, active site side chains in magenta, Ca2+ ions in green.8
Structure
The overall structure of the protein consists of 316 amino acid residues organized into two domains separated by the active site.12 The N-terminal domain is predominately composed of β-sheets, while the C-terminal domain is primarily composed of α-helices. The two large domains are separated by a central α-helix. One Zn2+ is bound via three residues in the active site within a conserved binding motif of HEXXH (His 142, His 146, Glu 166) located on the C-terminal side (Figure 2).14,15 The tetrahedral binding of Zn2+ is rounded out by a nucleophilic water molecule. This water molecule is capable of alternating between two distinct association positions, W1 and W2, which are in turn stabilized by His 231 and Glu 143, respectively.16 Four Ca2+ ions are associated with the enzyme that aide in thermostability.12
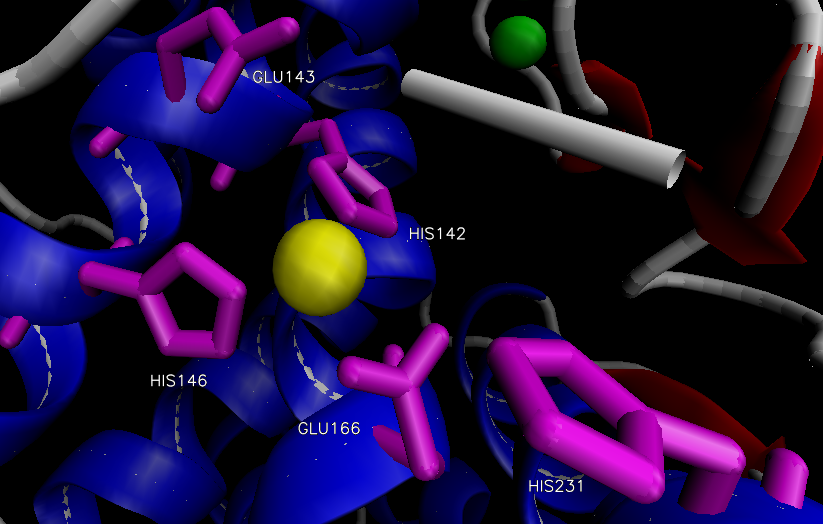
Figure 2. Active site of thermolysin.
Catalytic Residue Importance
Zinc: The bound Zn2+ is responsible for catalyzing peptide hydrolysis and stabilizing the various intermediates of the reaction. Although normally bound in a tetrahedral structure, during catalysis Zn2+ assumes a pentacoordinate geometry between the original three residues (His142, His146, and Glu166), the oxygen of the nucleophilic water, and the carbonyl oxygen of the substrate.7,14,17 The formation of a gem-diolate intermediate is stabilized by Zn2+.14,17 Removal of Zn2+ yields an inactive enzyme. Exogenous addition of other divalent transition metals, specifically Zn2+, Co2+, Fe2+, and Mn2+, results in the regaining of 100%, 200%, 60%, and 10% enzymatic activity.3 Zn2+ is also responsible for the polarization of the carbonyl bond of the substrate and the enhancement of nucleophilicity of the catalytic water molecule.
Glu143: Glu143 is responsible for the polarization of the catalytic water molecule leading to an enhancement of nucleophilicity. Additionally, Glu143 abstracts a proton from the water molecule and transfers it to the amide leaving group. In site-directed mutagenesis experiments involving the neutral protease of B. stearothermophilus, a protease with an amino acid sequence identical to that of thermolysin, Glu143Asp and Glu143Gln substitutions resulted in no catalytic activity of the enzyme.12,18 The mechanism proposed by Mock and colleagues suggests a diminished catalytic role of Glu143, which is instead used solely for charge stabilization with no association with the nucleophilic water molecule.19,20 Mutagenesis studies show that Glu143Asp substitutions result in inactive enzymes, despite the same side chain charge that would provide electrostatic stability.18
His231: His231 is responsible for substrate stabilization during hydrolysis. The carbonyl oxygen of the peptide is hydrogen bonded to N1 of His231 in the intermediate step of hydrolysis. Mutagenesis experiments involving His231Phe and His231Ala show 430- and 500-fold reductions in catalytic activity with no significant change in Km.21 These findings led to a proposed TLN-mechanism with an emphasis on the role of His231 as a general base, but this mechanism is less favored due to the residual activities of His231-substituted mutants compared to inactive mutants generated by Glu143 mutagenesis studies.18,19,21
Tyr157: The importance of Tyr157 has been debated in several mechanisms.18,22 Site-directed mutagenesis studies show an 80% drop in catalytic activity of Tyr157Trp mutants, and it has been suggested that the hydroxyl-H of Ty157 stabilizes the carboxylate-O of the peptide substrate during hydrolysis.17,23 Energetics modeling suggests a primary role for Tyr157 in transition state stabilization and substrate binding with an increase in the activation barrier energy of 2.7 kcal/mol and a decrease of binding affinity of 0.5 kcal/mol.24
Asp226: Asp226 has been suggested to stabilize the catalytic His231 through H-bonding of the Asp226 carboxylate to N3 of His231.17,22 Mutagenesis of Asp226Ala shows a relatively small decrease in catalytic activity of 40-fold and energetics modeling suggest an increase in overall ΔG‡ 2.2 kcal/mol.23,24
Mechanism
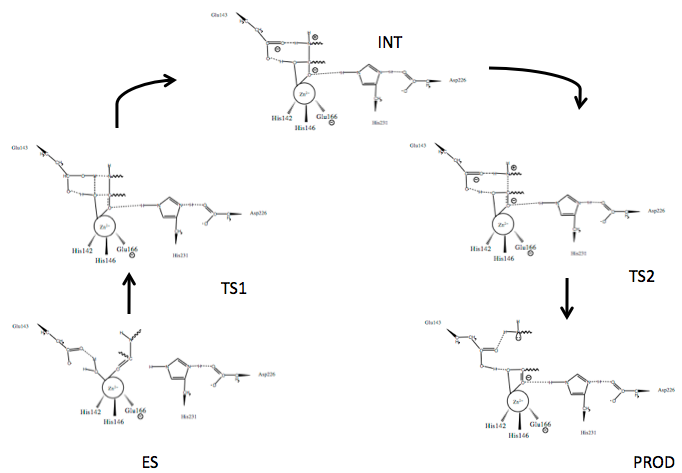
Scheme 1. Mechanism of peptide hydrolysis by thermolysin.24
Several mechanisms for TLN mediated peptide cleavage have been proposed with varying emphasis on the importance of the roles of the catalytic residues, Glu143 and His231, as well as several catalysis-associated residues, Tyr157 and Asp226. The generally accepted mechanism for TLN-mediated hydrolysis proceeds via the two-step process depicted in Scheme 1.24 Briefly:
- The active site houses the catalytic Zn2+ ion bound to three residues and a catalytic water (W)
- An incoming peptide substrate displaces the hydrogens of W1 towards Glu143, while the oxygen of W (OW) remains associated with Zn2+; Zn2+ associates with the carbonyl oxygen (OP) of the substrate forming the enzyme-substrate complex (ES)
- Polarizing actions on Zn2+ by Glu143 allow nucleophilic attack of carbonyl carbon (CP) by OW forming Transition State 1 (TS1); stabilized by Asp226 at N3, His231 N1 (originally protonated) stabilizes the substrate via hydrogen bonding at the carbonyl oxygen
- The intermediate (INT) gem-diolate is formed as Glu143 breaks the HW1-OW bond and the amide of the substrate (NP) hydrogen bonds HW2
- The NP-CP bond of the substrate begins to break as the CP-OP bond regains double bond character and the tetrahedral CP gem-diolate intermediate is pushed to Transition State 2 (TS2)
- The NP-CP is fully broken and the Glu143-bound HW2 bond begins to break as the new N-terminal hydrolyzed peptide is released (PROD)
- The active site is hydrated and is free for new peptide docking
The catalytic activity of TLN is dependent on both temperature and pH.25 The pKas of TLN are 5.0 and 8.25, and maximum catalytic activity has been measured at pH 7.2.25,26 The thermostability of TLN conferred by the four Ca2+ ions allows an increase in catalytic activity at temperatures approaching 40°C, with no significant loss in activity or conformation alteration until temperatures exceed 70°C.25,27
Inhibition and Inhibitors
Chelating agents such as EDTA have been shown to completely inactivate TLN and other metalloproteases by removal of the metal ion.3 In addition to direct removal of the catalytic metal ion, substrate and transition state analogs have been synthesized that greatly decrease the catalytic activity of TLN and TLN-like proteins. Transition state analogs that resemble the mechanistic transition states of the normal hydrolysis reaction catalyzed by TLN have been both isolated and synthesize. Phosphoramidates, a group of phosphoryl group containing amino acid or peptide compounds have been shown to mimic the tetrahedral intermediate of the TLN mechanism.28,29 Overall inhibition has been shown to vary from five to 1000-fold decrease in catalytic activity through use of transition state analogs.28,30 Holmquist and Vallee combined substrate analog inhibition with anionic ligands that tightly bind active site metals such as R-S- and R-P-O-.30 By combining two approaches, catalytic activity of TLN was decreased 10,000-fold. This compared to ~800-fold decrease when transition state analogs were combined with metal-binding ligands, suggesting a stronger inhibition effect of the substrate analog.30
Metalloexopeptidase: Carboxypeptidase A
Background
Carboxypeptidase A (carboxypolypeptidase; CPA; EC 3.4.17.1) is a 35 kD metalloenzyme within the zinc hydrolase family and as such contains a Zn2+ ion cofactor located within its active site.31,32 Originally isolated from bovine pancreas tissue in 1929, CPA is an exopeptidase that catalyzes the hydrolysis of C-terminal esters and peptides with large hydrophobic side chains.31,32 Biologically, CPA facilitates the breakdown of proteins during metabolism, while proposed commercial applications include its use the hydrolysis of cheese whey protein and the production of phenylalanine-free protein hydrolysates for use by individuals with phenylketonuria.33,34
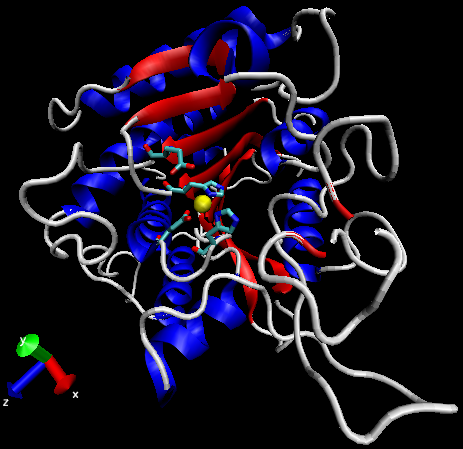
Figure 3: General structure of carboxypeptidase A (PDBID 1YME). Alpha-helices in blue, beta-sheets in red, Zn2+ ion in yellow, active site side chains in cyan.
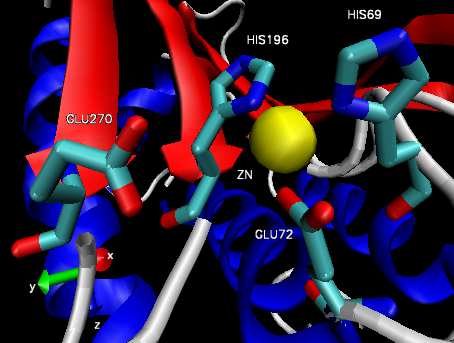
Figure 4: Carboxypeptidase A active site.32
Structure
The structure of CPA was first determined in 1967 using x-ray diffraction, making it one earliest proteins to have been characterized using the technique (Figure 3).35 CPA consists of a single chain containing 307 amino acids and a single Zn2+ ion in its active site.35 Zn2+ is stabilized within the active site through interactions with His69, Glu72, and His196 and is additionally bound by a catalytic water molecule that has been shown to interact with Glu270 (Figure 4).36 Mutagenesis studies have indicated that the additional residues Arg127, Tyr248, Arg71, Asn144, and Arg145 form an outer shell, which includes Glu270, around the active site and contribute to the catalytic function of CPA through the stabilization of substrate molecules.36 In both proposed mechanisms for the catalytic activity of CPA, Glu270 plays an important role, either acting as a general base-general acid or as a nucleophile.36
Mechanisms
Two mechanisms have been suggested for CPA catalyzed hydrolysis—the promoted-water pathway, also known as the general base-general acid pathway, and the nucleophilic, or anhydride, pathway—with experimental evidence existing for both mechanisms.36
Promoted-water pathway
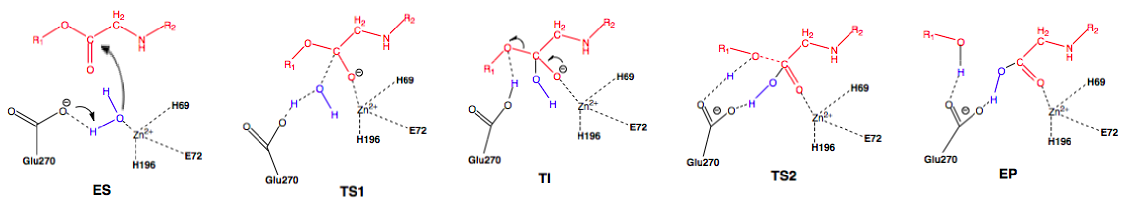
Scheme 2: Promoted-water (general base-general acid) pathway. Active site substituent side chains in black, substrate in red, water in blue.36
In the enzyme substrate (ES) complex, the Zn2+ tetrahedron consists of a water molecule, His69, Glu72, and His196 bound to the ion while the water molecule in the near attack position is maintained through hydrogen bonding with Glu270 and Ser197. The substrate position is maintained through interactions with Arg127, Asn144, Arg145, Tyr248, and Arg71. The water molecule attacks the scissile carbonyl carbon of the substrate molecule through nucleophilic addition with Glu270 acting as a general base, leading to the first transition state (TS1) and the formation of the tetrahedral intermediate (TI). Following the formation of the TI, the leaving group is protonated and the peptide bond is cleaved with Glu270 now severing as a general acid, forming the second transition state (TI2) and finally the enzyme product (EP) complex. An oxyanion hole, created by the polarization of the scissile carbonyl carbon of the substrate in the ES complex by Arg127 and the presence of Zn2+, helps stabilize the charge generated on the carbonyl oxygen during TS1 and TI.36
Nucleophilic pathway
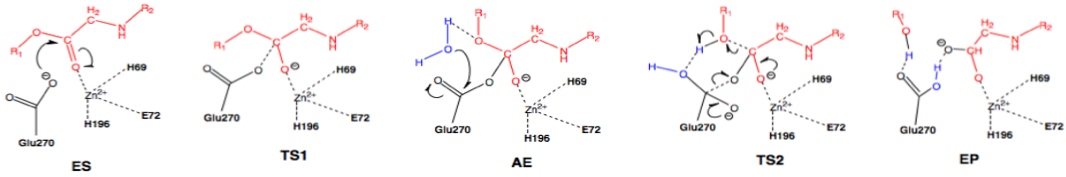
Scheme 3: Nucleophilic pathway. Active site substituent side chains in black, substrate in red, water in blue.36
In the enzyme substrate (ES) complex, the Zn2+ tetrahedron consists of His69, Glu72, His196, and the scissile carbonyl oxygen of the substrate molecule. This direct binding by Zn2+ polarizes the carbonyl oxygen, facilitating the nucleophilic attack by the carboxylate side chain of Glu270, which is in the near attack position. The substrate position is maintained through interactions with Arg127, Asn144, Arg145, Tyr248, and Arg71. The nucleophilic attack by Glu270 on the scissile carbonyl carbon leads to the first transition state (TS1) and the formation of the acyl enzyme intermediate (AE). A water molecule present in the active site nucleophilically attacks the carboxylate carbon of Glu270, resulting in deacylation and the transition through the second transition state (TS2) and finally the enzyme product (EP) complex.36
Computational evidence suggests that in regards to proteolysis, the promoted-water pathway is the only feasible pathway of the two. However, both pathways are feasible in esterolysis reactions, with the promoted-water pathway having the lower kinetic barrier. This suggests that under normal conditions the promoted-water pathway is favored, whereas at low temperatures, the formation of the tetrahedral intermediate by the promoter-water pathway and the formation of the acyl enzyme intermediate by the nucleophilic pathways are comparable. The second deacylation step in the nucleophilic pathway presents too high of a barrier however to be viable versus the promoted-water pathway. This then suggests that in regards to both proteolysis and esterolysis, the promoted-water pathway is the dominant pathway of CPA.36
Inhibitors and Inhibition
Ultraviolet-visible radiation (400 W, λ=250-750 nm) has been shown to cause uncompetitive inhibition of CPA with the decrease in enzymatic activity indirectly proportional to the irradiation time, with total enzymatic inactivation after 20 minutes of exposure. Additionally, exposure times of greater than 24 minutes are suspected to adversely affect the structure of CPA, resulting in the formation of protein aggregates.37
Active site-directed inhibitors of CPA, which are characterized by the presence of a terminal carboxylate, a hydrophobic side chain, and a zinc-binding group, have been identified, among which include the enantiomers of 2-benzyl-5-hydroxy-4-oxopentanoic acid. (L)-2-benzyl-5-hydroxy-4-oxopentanoic acid interacts through its carboxylate by forming hydrogen bonds with Arg145, Arg127, and Tyr248 and through its terminal hydroxyl group by forming a hydrogen bond with Ser197.38
Transition state analog inhibitors of CPA have also been found, which when bound with the enzyme active site create a pseudo-transition state complex. Both (R)-2-benzyl-3-nitropropanoic acid and (R)-2-benzyl-5-nitro-4-oxopentanoic acid are capable of inhibiting CPA by forming complexes with their respective nitro groups, Glu270, Arg127, and Zn2+.39,40
References
- Hooper, N.M. Families of zinc metalloproteases. FEBS Lett. 1994, 354, 1-6.
- Hase, C.C.; Finkelstein, R.A. Bacterial extracellular zinc-containing metalloproteases. Microbiol. Rev. 1993, 57, 823-837.
- Holmquist, B.; Vallee, B.L. Metal substitutions and inhibition of thermolysin: spectra of the cobalt enzyme. J. Biol. Chem. 1974, 249, 4601-4607.
- Vallee, B.L.; Auld, D.S. Zinc coordination, function, and structure of zinc enzymes and other proteins. Biochemistry. 1990, 29, 5647-5659.
- Barrett, A. J. Proteinases in Mammalian Cells and Tissues; Elsevier/North-Holland Biomedical Press, Amsterdam, Netherlands, 1977; p. 181-208.
- Garrett, R.H.; Grisham, C.M. Biochemistry; University of Virginia, United States, 2010; p. 103
- Lipscomb, W.N.; Strater, N. Recent advances in zinc enzymology. Chem. Rev. 1996, 96, 2375-2433.
- Hausrath, A.C.; Matthews, B.W. Thermolysin in the absence of substrate has an open conformation. Acta Crystallogr. 2002, 58, 1002-1007.
- Endo, S. Studies on protease produced by thermophilic bacteria. J. Ferment. Technol. 1962, 40, 346.
- Kooi, C.; Sokol, P.A. Differentiation of thermolysins and serralysins by monoclonal antibodies. J. Med. Microbiol. 1996, 45, 219-225.
- De Kreij, A.; Venema, G.; van den Burg, B. Substrate specificity in the highly heterogeneous M4 peptidase family is determined by a small subset of amino acids. J. Biol. Chem. 2000, 275, 31115-31120.
- Titani, K.; Hermodson, M.A.; Ericsson, L.H.; Walsh, K.A.; Neurath, H. Amino-acid sequence of thermolysin. Nat. New Biol. 1972, 238, 35-37.
- Ooshima, H.; Mori, H.; Harano, Y. Synthesis of aspartame precursor by thermolysin solid in organic solvent. Biotechnol. Lett. 1985, 7, 789-792.
- Matthews, B.W.; Jansonius, J.N.; Colman, P.M.; Schoenborn, B.P.; Dupourque, D. Three-dimensional structure of thermolysin. Nat. New Biol. 1972, 238, 37-41.
- Rawlings, N.D.; Barrett, A.J. Evolutionary families of peptidases. Biochem. J. 1993, 290, 205-218.
- Holland, D.R.; Hausrath, A.C.; Juers, D.; Matthews, B.W. Structural analysis of zinc substitutions in the active site of thermolysin. Protein Sci. 1995, 4, 1955-1965
- Hangauer, D.G.; Monzingo, A.F.; Matthews, B.W. An interactive computer graphics study of thermolysin-catalyzed peptide cleavage and inhibition by N-carboxymethyl peptides. Biochemistry. 1984, 23, 5730-5741.
- Kubo, M.; Mitsuda, Y.; Takagi, M.; Imanaka, T. Alteration of specific activity and stability of thermostable neutral protease by site-directed mutagenesis. Appl. Environ. Microbiol. 1992, 58, 3779-3783.
- Mock, W.L.; Aksamawati, M. Binding to thermolysin of phenolate-containing inhibitors necessitates a revised mechanism of catalysis. Biochem. J. 1994, 302, 57-68.
- Mock, W.L., Stanford, D.J. Arazoformyl dipeptide substrates for thermolysin: confirmation of a reverse protonation catalytic mechanism. Biochemistry. 1996, 35, 7369-7377.
- Beaumont, A.; O’Donohue, M.J.; Paredes, N.; Rousselet, N.; Assicot, M.; Buhuon, C.; Fournie-Zaluski, M.; Roques, B.P. The role of histidine 231 in thermolysin-like enzymes. J. Biol. Chem. 1995, 270, 16803-16808.
- Holden, H.M.; Matthews, B.W. The binding of L-valyl-l-tryptophan to crystalline thermolysin illustrates the mode of interaction of a product peptide hydrolysis. J. Biol. Chem. 1988, 263, 3256-3260.
- Marie-Claire, C.; Ruffet, E.; Tiraboschi, G.; Fournie-Zaluski, M. Differences in transition state stabilization between thermolysin (E.C. 3.4.24.27) and neprilysin (E.C. 3.4.24.11). FEBS Lett. 1998, 438, 215-219.
- Pelmenschikov, V.; Blomberg, M.R.A.; Siegbahn, P.E.M. A theoretical study of the mechanism for peptide hydrolysis by thermolysin. J. Biol. Inorg. Chem. 2002, 7, 284-298.
- Kunugi, S.; Hirohara, H.; Ise, N. pH and temperature dependences of thermolysin catalysis: catalytic role of zinc-coordinated water. Eur. J. Biochem. 1982, 124, 157-163.
- Feder, J.; Brougham, L.R.; Wildi, B.S. Inhibition of thermolysin by dipeptides. Biochemistry. 1974, 13, 1186-1189.
- Ohta, Y.; Ogura, Y.; Wada, A. Thermostable protease from thermophilic bacteria I: thermostability, physiochemical properties, and amino acid composition. J. Biol. Chem. 1966, 241, 5919-5925.
- Kam, C.; Nishino, N.; Powers, J.C. Inhibition of thermolysin and carboxypeptidase A by phosphoramidates. Biochemistry. 1979, 18, 3032-3038.
- Suda, H.; Aoyagi, T.;Takeuchi, T; Umezawa, H. Antipain, a new protease inhibitor isolated form actiomycetes. J. Antibiot. 1972, 25, 263-266.
- Holmquist, B.; Vallee, B.L. Metal-coordinating substrate analogs as inhibitors of metalloenzymes. Proc. Natl. Acad. Sci. USA. 1979, 76, 6216-6220.
- Kilshtain, A.V.; Warshel, A. On the origin of the catalytic power of carboxypeptidase A and other metalloenzymes. Proteins 2009, 77, 536-550.
- Szeto, M.W.Y.; Mujika, J.I.; Zurek, J.; Mulholland, A.J.; Harvey, J.N. QM/MM study on the mechanism of peptide hydrolysis by carboxypeptidase A. J. Mol. Struc.-THEOCHEM 2008, 898, 106-114.
- Pinto, G.A.; Tardioli, P.W.; Cabrera-Padilla, R.Y.; Galvão, C.M.A.; Giordano, R.C.; Giordano, R.L.C. Amino acid yields during proteolysis catalyzed by carboxypeptidase A are strongly dependent on substrate pre-hydrolysis. Biochem. Eng. J. 2008, 39, 328-337.
- Xu, D.; Guo, H. Quantum Mechanical/Molecular Mechanical and Density Functional Theory Studies of a Prototypical Zinc Peptidase (Carboxypeptidase A) Suggest a General Acid-General Base Mechanism. J. Am. Chem. Soc. 2009, 131, 9780-9788.
- Quicho, F.A.; McMurray, C.H.; Lipscomb, W.N. Similarities Between the Conformation of Arsanilazotyrosine 248 of Carboxypeptidase Aa in the Crystalline State and in Solution. Proc. Nat. Acad. Sci. USA 1972, 69, 2850-2854.
- Wu, S.; Zhang, C.; Xu, D.; Guo, H. Catalysis of Carboxypeptidase A: Promoted-Water versus Nucleophilic Pathways. J. Phys. Chem. B 2010, 114, 9259-9267.
- Ibarz, A.; Garvín, A.; Garza, S.; Pagán, J. Inactivation of carboxypeptidase A and trypsin by UV-visible light. Innov. Food Sci. Emerg. 2009, 10, 517-521.
- Wang, S.F.; Jin, J.Y.; Zeng, Z.H.; Tian, G.R. Optical 2-benzyl-5-hydroxy-4-oxopentanoic acids against carboxypeptidase A: Synthesis, kinetic evaluation and X-ray crystallographic study. Chinese Chem. Lett. 2010, 21, 159-162.
- Wang, S.H.; Wang, S.F.; Xuan, W.; Zeng, Z.H.; Yin, J.Y.; Ma, J.; Tian, G.R. Nitro as a novel zinc-binding group in the inhibition of carboxypeptidase A. Bioorgan. Med. Chem. 2008, 16, 3596-3601.
- Wang, S.F.; Tian, G.R.; Zhang, W.Z.; Jin, J.Y. Characterization of a a-nitromethyl ketone as a new zinc-binding group based on structural analysis of its complex with carboxypeptidase A. Bioorg. Med. Chem. Lett. 2009, 19, 5009-5011.
Contributors and Attributions
- Hank Kimbrough (Truman State University)
- Christopher Tracy (Truman State University)