
7.6: Classifying Separation Techniques
- Page ID
- 162885

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)We can separate an analyte and an interferent if there is a significant difference in at least one of their chemical or physical properties. Table 7.6.1 provides a partial list of separation techniques, organized by the chemical or physical property affecting the separation.
| basis of separation | separation technique(s) |
|---|---|
| size | filtration; dialaysis; size-exclusion chromatography |
| mass or density | centrifugation |
| complex formation | masking |
| change in physical state | distillation; sublimation; recrystalization |
| change in chemical state | precipitation; electrodeposition; volatilization |
| partitioning between phases | extraction; chromatography |
Separations Based on Size
Size is the simplest physical property we can exploit in a separation. To accomplish the separation we use a porous medium through which only the analyte or the interferent can pass. Examples of size-based separations include filtration, dialysis, and size-exclusion.
In a filtration we separate a particulate interferent from soluble analytes using a filter with a pore size that will retain the interferent. The solution that passes through the filter is called the filtrate, and the material retained by the filter is the retentate. Gravity filtration and suction filtration using filter paper are techniques with which you should already be familiar. A membrane filter is the method of choice for particulates that are too small to be retained by filter paper. Figure 7.6.1 provides information about three types of membrane filters. For applications of gravity filtration and suction filtration in gravimetric methods of analysis, see Chapter 8.
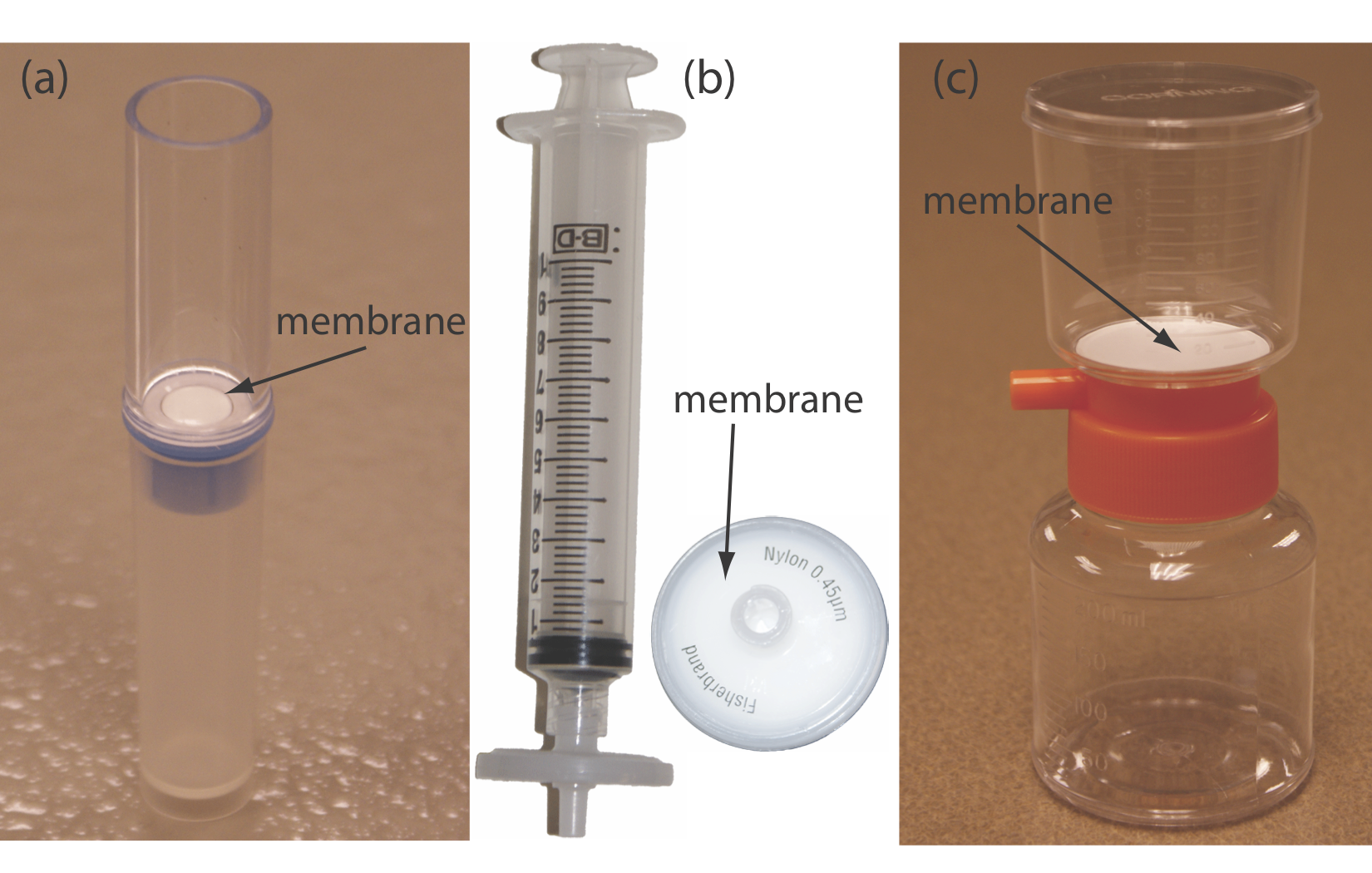
Dialysis is another example of a separation technique in which size is used to separate the analyte and the interferent. A dialysis membrane usually is made using cellulose and fashioned into tubing, bags, or cassettes. Figure 7.6.2 shows an example of a commercially available dialysis cassette. The sample is injected into the dialysis membrane, which is sealed tightly by a gasket, and the unit is placed in a container filled with a solution with a composition different from the sample. If there is a difference in a species’ concentration on the membrane’s two sides, the resulting concentration gradient provides a driving force for its diffusion across the membrane. While small species freely pass through the membrane, larger species are unable to pass. Dialysis frequently is used to purify proteins, hormones, and enzymes. During kidney dialysis, metabolic waste products, such as urea, uric acid, and creatinine, are removed from blood by passing it over a dialysis membrane.

Size-exclusion chromatography is a third example of a separation technique that uses size as a means to effect a separation. In this technique a column is packed with small, approximately 10-μm, porous polymer beads of cross-linked dextrin or polyacrylamide. The pore size of the particles is controlled by the degree of cross-linking, with more cross-linking producing smaller pore sizes. The sample is placed into a stream of solvent that is pumped through the column at a fixed flow rate. Those species too large to enter the pores pass through the column at the same rate as the solvent. Species that enter into the pores take longer to pass through the column, with smaller species requiring more time to pass through the column. Size-exclusion chromatography is widely used in the analysis of polymers, and in biochemistry, where it is used for the separation of proteins. A more detailed treatment of size-exclusion chromatography, which also is called gel permeation chromatography, is in Chapter 12.
Separations Based on Mass or Density
If the analyte and the interferent have different masses or densities, then a separation using centrifugation may be possible. The sample is placed in a centrifuge tube and spun at a high angular velocity, measured in revolutions per minute (rpm). The sample’s constituents experience a centrifugal force that pulls them toward the bottom of the centrifuge tube. Those species that experience the greatest centrifugal force have the fastest sedimentation rate and are the first to reach the bottom of the centrifuge tube. If two species have the same density, their separation is based on a difference in mass, with the heavier species having the greater sedimentation rate. If the species are of equal mass, then the species with the larger density has the greatest sedimentation rate.
Centrifugation is an important separation technique in biochemistry. Table 7.6.2 , for example, lists conditions for separating selected cellular components. We can separate lysosomes from other cellular components by several differential centrifugations, in which we divide the sample into a solid residue and a supernatant solution. After destroying the cells, the solution is centrifuged for 20 minutes at \(15000 \times g\) (a centrifugal force that is 15 000 times the earth’s gravitational force), leaving a solid residue of cell membranes and mitochondria. The supernatant, which contains the lysosomes, is isolated by decanting it from the residue and then centrifuged for 30 minutes at \(30000 \times g\), leaving a solid residue of lysosomes. Figure 7.6.3 shows a typical centrifuge capable of producing the centrifugal forces needed for biochemical separations.
| components | centrifugal force (\(\times g\)) | time (min) |
|---|---|---|
| eukaryotic cells | 1000 | 5 |
| cell membranes; nuclei | 4000 | 10 |
| mitochondria, bacterial cells | 15000 | 20 |
| lysosomes; bacterial membranes | 30000 | 30 |
| ribosomes | 100000 | 180 |
|
Source: Adapted from Zubay, G. Biochemistry, 2nd ed. Macmillan: New York, 1988, p.120. |
||

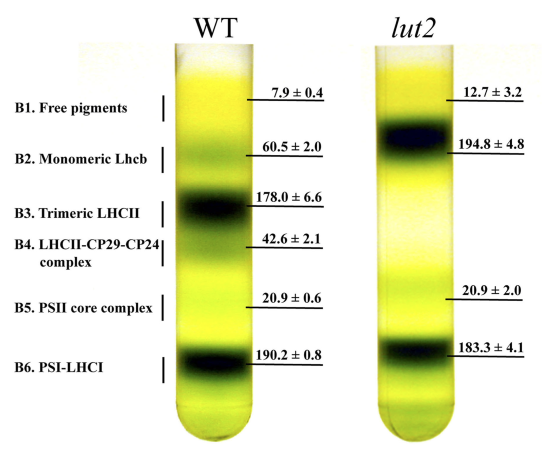
An alternative approach to differential centrifugation is a density gradient centrifugation. To prepare a sucrose density gradient, for example, a solution with a smaller concentration of sucrose—and, thus, of lower density—is gently layered upon a solution with a higher concentration of sucrose. Repeating this process several times, fills the centrifuge tube with a multi-layer density gradient. The sample is placed on top of the density gradient and centrifuged using a force greater than \(150000 \times g\). During centrifugation, each of the sample’s components moves through the gradient until it reaches a position where its density matches the surrounding sucrose solution. Each component is isolated as a separate band positioned where its density is equal to that of the local density within the gradient. Figure 7.6.4 provides an example of a typical sucrose density centrifugation for separating plant thylakoid membranes.
Separations Based on Complexation Reactions (Masking)
One widely used technique for preventing an interference is to bind the interferent in a strong, soluble complex that prevents it from interfering in the analyte’s determination. This process is known as masking. As shown in Table 7.6.3 , a wide variety of ions and molecules are useful masking agents, and, as a result, selectivity is usually not a problem.
Technically, masking is not a separation technique because we do not physically separate the analyte and the interferent. We do, however, chemically isolate the interferent from the analyte, resulting in a pseudo-separation.
| masking agent | elements whose ions are masked |
|---|---|
| CN– |
Ag, Au, Cd, Co, Cu, Fe, Hg, Mn, Ni, Pd, Pt, Zn |
| SCN– |
Ag, Cd, Co, Cu, Fe, Ni, Pd, Pt, Zn |
| NH3 |
Ag, Co, Ni, Cu, Zn |
| F– | Al, Co, Cr, Mg, Mn, Sn, Zn |
| \(\text{S}_2\text{O}_3^{2-}\) |
Au, Ce, Co, Cu, Fe, Hg, Mn, Pb, Pd, Pt, Sb, Sn, Zn |
| tartrate | Al, Ba, Bi, Ca, Ce, Co, Cr, Cu, Fe, Hg, Mn, Pb, Pd, Pt, Sb, Sn, Zn |
| oxalate |
Al, Fe, Mg, Mn |
| thioglycolic acid |
Cu, Fe, Sn |
|
Source: Meites, L. Handbook of Analytical Chemistry, McGraw-Hill: New York, 1963. |
|
Using Table 7.6.3 , suggest a masking agent for the analysis of aluminum in the presence of iron.
Solution
A suitable masking agent must form a complex with the interferent, but not with the analyte. Oxalate, for example, is not a suitable masking agent because it binds both Al and Fe. Thioglycolic acid, on the other hand, is a selective masking agent for Fe in the presence of Al. Other acceptable masking agents are cyanide (CN–) thiocyanate (SCN–), and thiosulfate (\(\text{S}_2\text{O}_3^{2-}\)).
Using Table 7.6, suggest a masking agent for the analysis of Fe in the presence of Al.
- Answer
-
The fluoride ion, F–, is a suitable masking agent as it binds with Al3+ to form the stable \(\text{AlF}_6^{3-}\) complex, leaving iron in solution.
As shown in Example 7.6.2 , we can judge a masking agent’s effectiveness by considering the relevant equilibrium constants.
Show that CN– is an appropriate masking agent for Ni2+ in a method where nickel’s complexation with EDTA is an interference.
Solution
The relevant reactions and formation constants are
\[\mathrm{Ni}^{2+}(a q)+\mathrm{Y}^{4-}(a q)\rightleftharpoons \mathrm{NiY}^{2-}(a q) \quad K_{1}=4.2 \times 10^{18} \nonumber\]
\[\mathrm{Ni}^{2+}(a q)+4 \mathrm{CN}^{-}(a q)\rightleftharpoons \mathrm{Ni}(\mathrm{CN})_{4}^{2-}(a q) \quad \beta_{4}=1.7 \times 10^{30} \nonumber\]
where Y4– is an abbreviation for EDTA. Cyanide is an appropriate masking agent because the formation constant for \(\text{Ni(CN)}_4^{2-}\) is greater than that for the Ni–EDTA complex. In fact, the equilibrium constant for the reaction in which EDTA displaces the masking agent
\[\mathrm{Ni}(\mathrm{CN})_{4}^{2-}(a q)+\mathrm{Y}^{4-}(a q) \rightleftharpoons \mathrm{NiY}^{2-}(a q)+4 \mathrm{CN}^{-}(a q) \nonumber\]
\[K=\frac{K_{1}}{\beta_{4}}=\frac{4.2 \times 10^{18}}{1.7 \times 10^{30}}=2.5 \times 10^{-12} \nonumber\]
is sufficiently small that \(\text{Ni(CN)}_4^{2-}\) is relatively inert in the presence of EDTA.
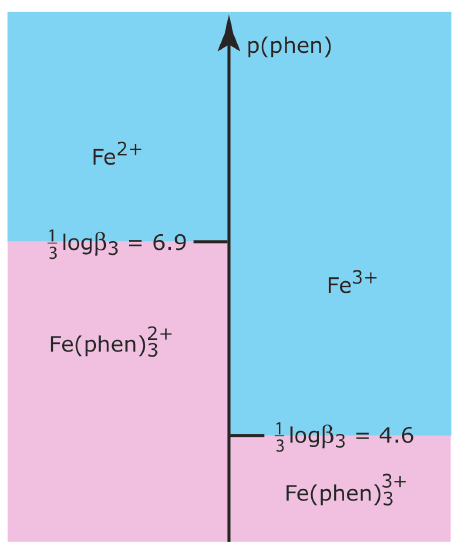
Use the formation constants in Appendix 12 to show that 1,10-phenanthroline is a suitable masking agent for Fe2+ in the presence of Fe3+. Use a ladder diagram to define any limitations on using 1,10-phenanthroline as a masking agent. See Chapter 6 for a review of ladder diagrams.
- Answer
-
The relevant reactions and equilibrium constants are
\[\begin{array}{ll}{\mathrm{Fe}^{2+}(a q)+3 \mathrm{phen}(a q)} & {\rightleftharpoons\mathrm{Fe}(\mathrm{phen})_{3}^{2+}(a q) \quad \beta_{3}=5 \times 10^{20}} \\ {\mathrm{Fe}^{3+}(a q)+3 \mathrm{phen}(a q)} & {\rightleftharpoons \mathrm{Fe}(\mathrm{phen})_{3}^{3+}(a q) \quad \beta_{3}=6 \times 10^{13}}\end{array} \nonumber\]
where phen is an abbreviation for 1,10-phenanthroline. Because \(\beta_3\) is larger for the complex with Fe2+ than it is for the complex with Fe3+,1,10-phenanthroline will bind Fe2+ before it binds Fe3+. A ladder diagram for this system (as shown below) suggests that an equilibrium p(phen) between 5.6 and 5.9 will fully complex Fe2+ without any significant formation of the \(\text{Fe(phen)}_3^{3+}\) complex. Adding a stoichiometrically equivalent amount of 1,10-phenanthroline to a solution of Fe2+ is sufficient to mask Fe2+ in the presence of Fe3+. A large excess of 1,10-phenanthroline, however, decreases p(phen) and allows for the formation of both metal–ligand complexes.
Separations Based on a Change of State
Because an analyte and its interferent are usually in the same phase, we can achieve a separation if one of them undergoes a change in its physical state or its chemical state.
Changes in Physical State
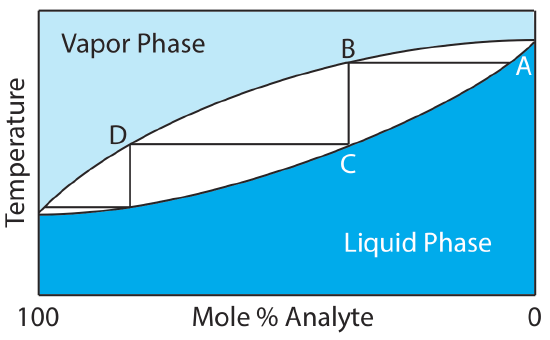
When the analyte and the interferent are miscible liquids, separation by distillation is possible if their boiling points are significantly different. Figure 7.6.5 shows the progress of a distillation as a plot of temperature versus the composition of mixture’s vapor-phase and liquid-phase. The initial liquid mixture (point A), contains more interferent than analyte. When this solution is brought to its boiling point, the vapor phase in equilibrium with the liquid phase is enriched in analyte (point B). The horizontal line that connects points A and B represents this vaporization equilibrium. Condensing the vapor phase at point B, by lowering the temperature, creates a new liquid phase with a composition identical to that in the vapor phase (point C). The vertical line that connects points B and C represents this condensation equilibrium. The liquid phase at point C has a lower boiling point than the original mixture, and is in equilibrium with the vapor phase at point D. This process of repeated vaporization and condensation gradually separates the analyte and the interferent.
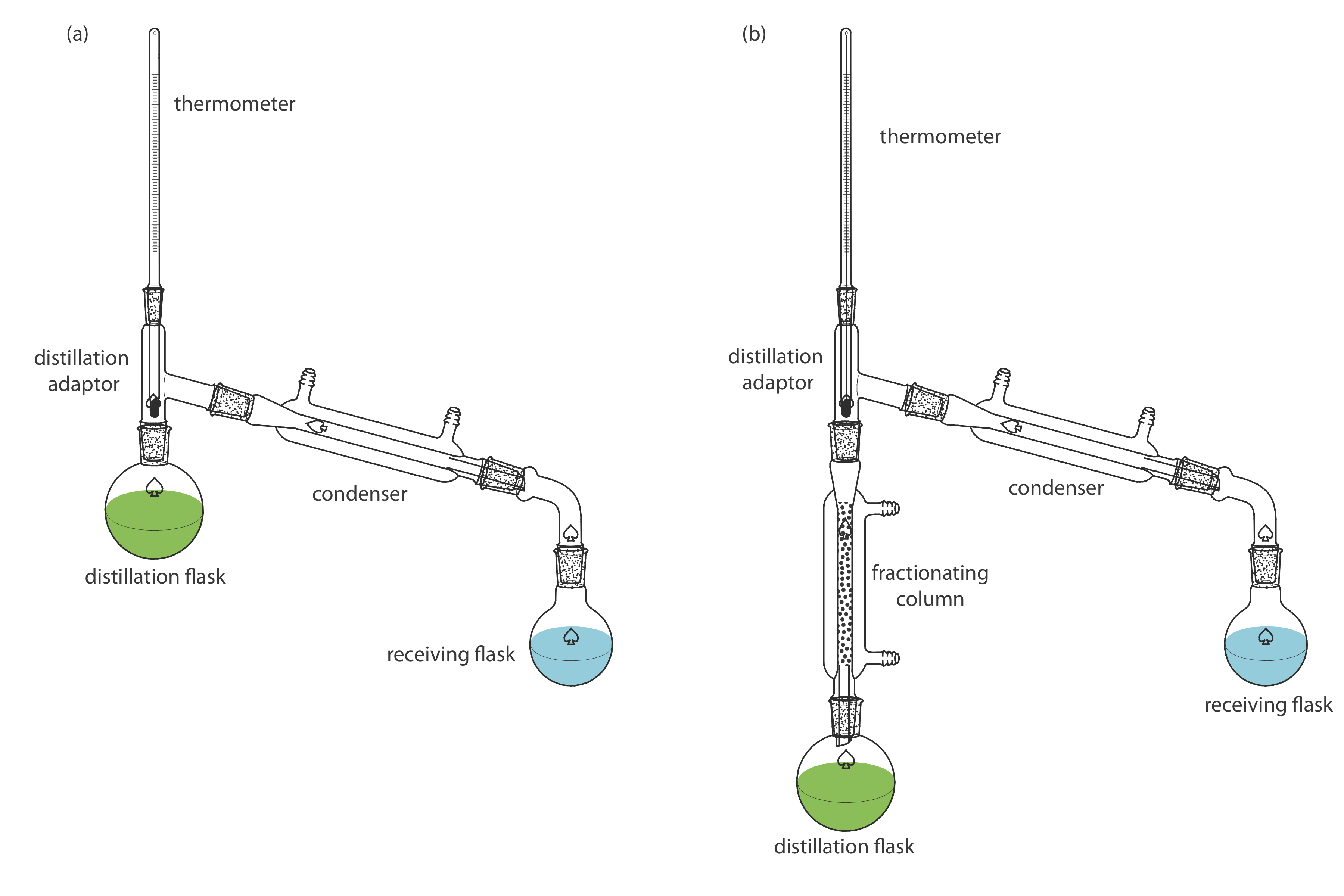
Two experimental set-ups for distillations are shown in Figure 7.6.6 . The simple distillation apparatus shown in Figure 7.6.6 a is useful only for separating a volatile analyte (or interferent) from a non-volatile interferent (or analyte), or for separating an analyte and an interferent whose boiling points differ by more than 150oC. A more efficient separation is achieved using the fractional distillation apparatus in Figure 7.6.6 b. Packing the fractionating column with a high surface area material, such as a steel sponge or glass beads, provides more opportunity for the repeated process of vaporization and condensation necessary to effect a complete separation.
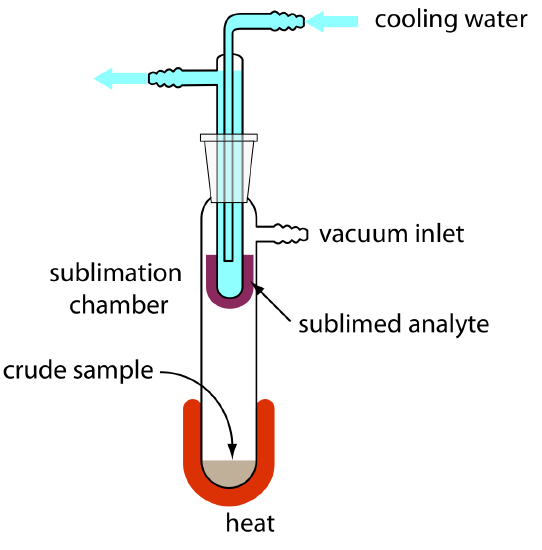
When the sample is a solid, sublimation may provide a useful separation of the analyte and the interferent. The sample is heated at a temperature and pressure below the analyte’s triple point, allowing it to vaporize without passing through a liquid state. Condensing the vapor recovers the purified analyte (Figure 7.6.7 ). A useful analytical example of sublimation is the isolation of amino acids from fossil mollusk shells and deep-sea sediments [Glavin, D. P.; Bada, J. L. Anal. Chem. 1998, 70, 3119–3122].
Recrystallization is another method for purifying a solid. A solvent is chosen in which the analyte’s solubility is significant when the solvent is hot and minimal when the solvent is cold. The interferents must be less soluble in the hot solvent than the analyte or present in much smaller amounts. After heating a portion of the solvent in an Erlenmeyer flask, small amounts of sample are added until undissolved sample is visible. Additional hot solvent is added until the sample redissolves, or until only insoluble impurities remain. This process of adding sample and solvent is repeated until the entire sample is added to the Erlenmeyer flask. Any insoluble impurities are removed by filtering the hot solution. The solution is allowed to cool slowly, which promotes the growth of large, pure crystals, and then cooled in an ice bath to minimize solubility losses. The purified sample is isolated by filtration and rinsed to remove any soluble impurities. Finally, the sample is dried to remove any remaining traces of the solvent. Further purification, if necessary, is accomplished by additional recrystallizations.
Changes in Chemical State
Distillation, sublimation, and recrystallization use a change in physical state to effect a separation. Chemical reactivity also is a useful tool for separating analytes and interferents. For example, we can separate SiO2 from a sample by reacting it with HF to form SiF4. Because SiF4 is volatile, it is easy to remove by evaporation. If we wish to collect the reaction’s volatile product, then a distillation is possible. For example, we can isolate the \(\text{NH}_4^+\) in a sample by making the solution basic and converting it to NH3. The ammonia is then removed by distillation. Table 7.6.4 provides additional examples of this approach for isolating inorganic ions.
| analyte | treatment | isolated species |
|---|---|---|
| \(\text{CO}_3^{2-}\) | \(\mathrm{CO}_{3}^{2-}(a q)+2 \mathrm{H}_{3} \mathrm{O}^{+}(a q) \rightarrow \mathrm{CO}_{2}(g)+3 \mathrm{H}_{2} \mathrm{O}(l)\) | CO2 |
| \(\text{NH}_4^+\) | \(\mathrm{NH}_{4}^{+}(a q)+\mathrm{OH}^{-}(a q) \rightarrow \mathrm{NH}_{3}(a q)+\mathrm{H}_{2} \mathrm{O}(l)\) | NH3 |
| \(\text{SO}_3^-\) | \(\mathrm{SO}_{3}^{2-}(a q)+2 \mathrm{H}_{3} \mathrm{O}^{+}(a q) \rightarrow \mathrm{SO}_{2}(g)+3 \mathrm{H}_{2} \mathrm{O}(l)\) | SO2 |
| S2– | \(\text{S}^{2-}(a q)+2 \text{H}_{3} \mathrm{O}^{+}(a q) \rightarrow \text{H}_{2} \text{S}(g)+2 \text{H}_{2} \text{O}(l)\) | H2S |
Another reaction for separating analytes and interferents is precipitation. Two important examples of using a precipitation reaction in a separation are the pH-dependent solubility of metal oxides and hydroxides, and the pH-dependent solubility of metal sulfides.
Separations based on the pH-dependent solubility of oxides and hydroxides usually use a strong acid, a strong base, or an NH3/NH4Cl buffer to adjust the pH. Most metal oxides and hydroxides are soluble in hot concentrated HNO3, although a few oxides, such as WO3, SiO2, and SnO2 remain insoluble even under these harsh conditions. To determine the amount of Cu in brass, for example, we can avoid an interference from Sn by dissolving the sample with a strong acid and filtering to remove the solid residue of SnO2.
Most metals form a hydroxide precipitate in the presence of concentrated NaOH. Those metals that form amphoteric hydroxides, however, do not precipitate because they react to form higher-order hydroxo-complexes. For example, Zn2+ and Al3+ do not precipitate in concentrated NaOH because they form the soluble complexes \(\text{Zn(OH)}_3^-\) and \(\text{Al(OH)}_4^-\). The solubility of Al3+ in concentrated NaOH allows us to isolate aluminum from impure samples of bauxite, an ore of Al2O3. After crushing the ore, we place it in a solution of concentrated NaOH, dissolving the Al2O3 and forming \(\text{Al(OH)}_4^-\). Other oxides in the ore, such as Fe2O3 and SiO2, remain insoluble. After filtering, we recover the aluminum as a precipitate of Al(OH)3 by neutralizing some of the OH– with acid.
The pH of an NH3/NH4Cl buffer (pKa = 9.26) is sufficient to precipitate most metals as the hydroxide. The alkaline earths and alkaline metals, however, do not precipitate at this pH. In addition, metal ions that form soluble complexes with NH3, such as Cu2+, Zn2+, Ni2+, and Co2+ also do not precipitate under these conditions.
The use of S2– as a precipitating reagent is one of the earliest examples of a separation technique. In Fresenius’s 1881 text A System of Instruction in Quantitative Chemical Analysis, sulfide frequently is used to separate metal ions from the remainder of the sample’s matrix [Fresenius. C. R. A System of Instruction in Quantitative Chemical Analysis, John Wiley and Sons: New York, 1881]. Sulfide is a useful reagent for separating metal ions for two reasons: (1) most metal ions, except for the alkaline earths and alkaline metals, form insoluble sulfides; and (2) these metal sulfides show a substantial variation in solubility. Because the concentration of S2– is pH-dependent, we can control which metal ions precipitate by adjusting the pH. For example, in Fresenius’s gravimetric procedure for the determination of Ni in ore samples (see Figure 1.1.1 for a schematic diagram of this procedure), sulfide is used three times to separate Co2+ and Ni2+ from Cu2+ and, to a lesser extent, from Pb2+.
Separations Based on a Partitioning Between Phases
The most important group of separation techniques uses a selective partitioning of the analyte or interferent between two immiscible phases. If we bring a phase that contains the solute, S, into contact with a second phase, the solute will partition itself between the two phases, as shown by the following equilibrium reaction.
\[S_{\text { phase } 1} \rightleftharpoons S_{\text { phase } 2} \label{7.1}\]
The equilibrium constant for reaction \ref{7.1}
\[K_{\mathrm{D}}=\frac{\left[S_{\mathrm{phase} \ 2}\right]}{\left[S_{\mathrm{phase} \ 1}\right]} \nonumber\]
is called the distribution constant or the partition coefficient. If KD is sufficiently large, then the solute moves from phase 1 to phase 2. The solute will remain in phase 1 if the partition coefficient is sufficiently small. When we bring a phase that contains two solutes into contact with a second phase, a separation of the solutes is possible if KD is favorable for only one of the solutes. The physical states of the phases are identified when we describe the separation process, with the phase that contains the sample listed first. For example, if the sample is in a liquid phase and the second phase is a solid, then the separation involves liquid–solid partitioning.
Extraction Between Two Phases
We call the process of moving a species from one phase to another phase an extraction. Simple extractions are particularly useful for separations where only one component has a favorable partition coefficient. Several important separation techniques are based on a simple extraction, including liquid–liquid, liquid–solid, solid–liquid, and gas–solid extractions.
Liquid-Liquid Extractions
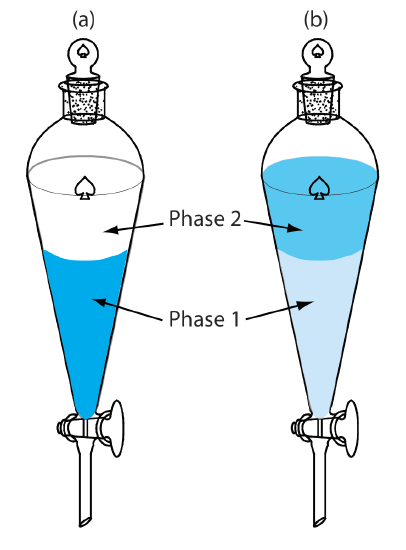
A liquid–liquid extraction usually is accomplished using a separatory funnel (Figure 7.6.8 ). After placing the two liquids in the separatory funnel, we shake the funnel to increase the surface area between the phases. When the extraction is complete, we allow the liquids to separate. The stopcock at the bottom of the separatory funnel allows us to remove the two phases.
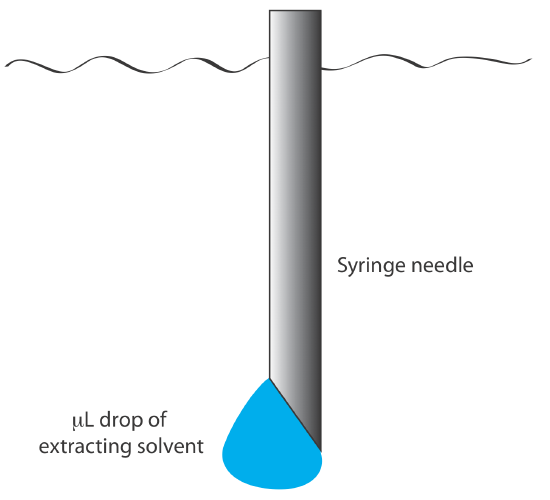
We also can carry out a liquid–liquid extraction without a separatory funnel by adding the extracting solvent to the sample’s container. Pesticides in water, for example, are preserved in the field by extracting them into a small volume of hexane. A liquid–liquid microextraction, in which the extracting phase is a 1-µL drop suspended from a microsyringe (Figure 7.6.9 ), also has been described [Jeannot, M. A.; Cantwell, F. F. Anal. Chem. 1997, 69, 235–239]. Because of its importance, a more thorough discussion of liquid–liquid extractions is in Chapter7.7.
Solid Phase Extractions
In a solid phase extraction of a liquid sample, we pass the sample through a cartridge that contains a solid adsorbent, several examples of which are shown in Figure 7.6.10 . The choice of adsorbent is determined by the species we wish to separate. Table 7.6.5 provides several representative examples of solid adsorbents and their applications.

| absorbent | structure | properties and uses |
|---|---|---|
| silica | 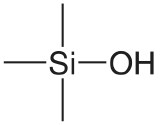 |
|
| aminopropyl | 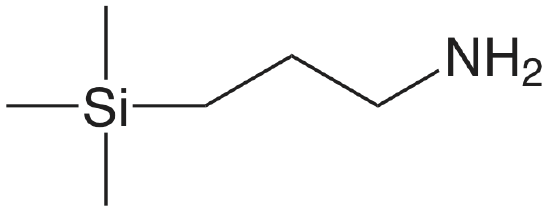 |
|
| cyanopropyl | 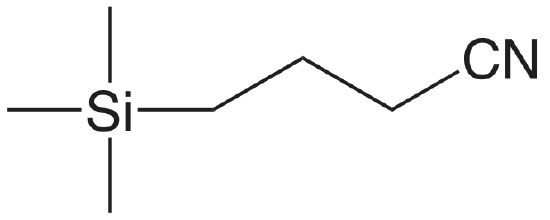 |
|
| diol | 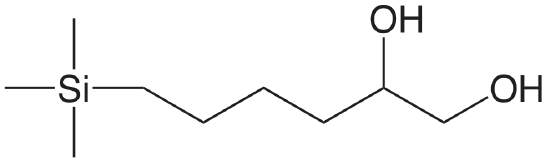 |
|
| octadecyl (C–18) | —C18H37 |
|
| octyl (C–8) | —C8H17 |
|
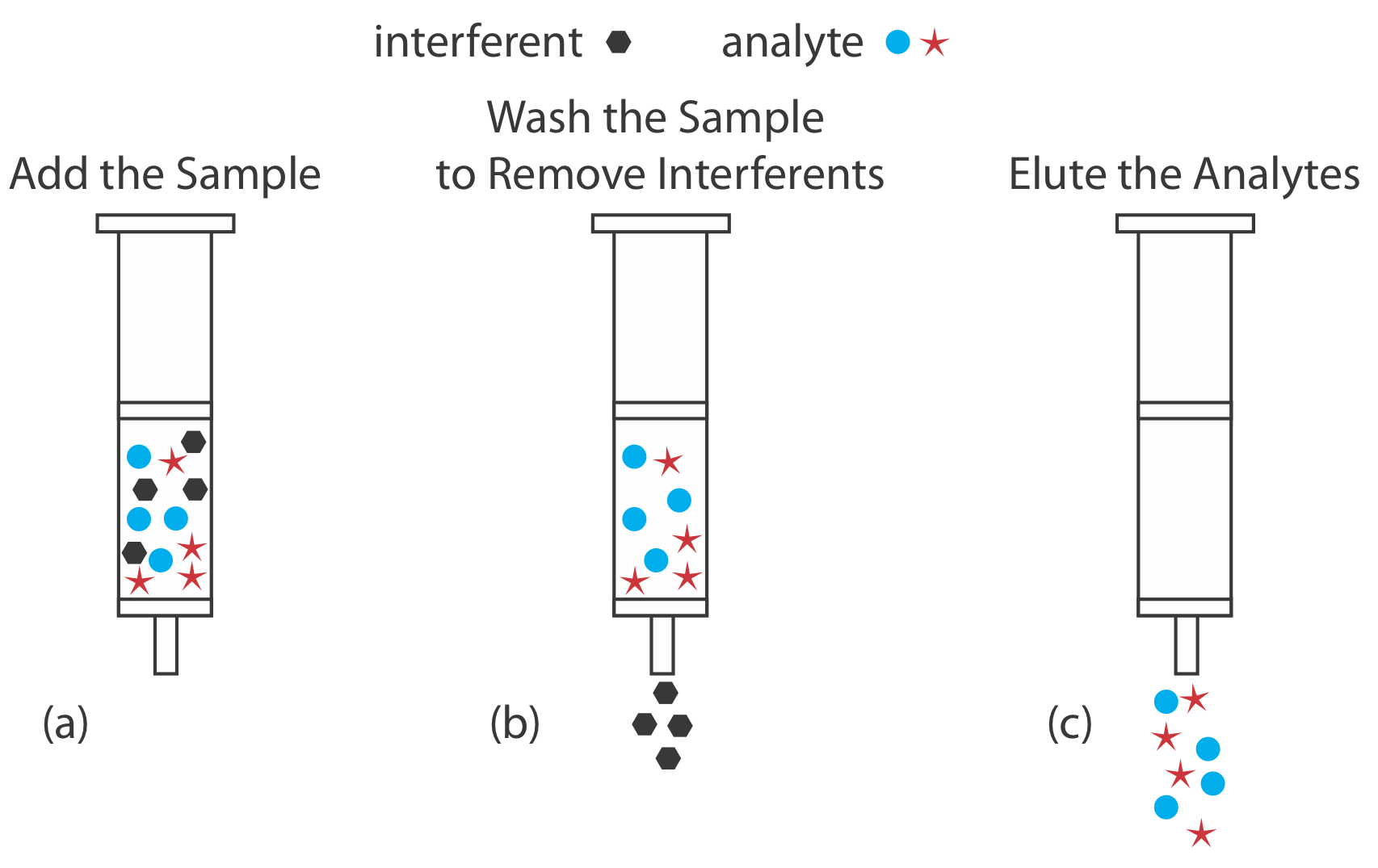
As an example, let’s examine a procedure for isolating the sedatives secobarbital and phenobarbital from serum samples using a C-18 solid adsorbent [Alltech Associates Extract-Clean SPE Sample Preparation Guide, Bulletin 83]. Before adding the sample, the solid phase cartridge is rinsed with 6 mL each of methanol and water. Next, a 500-μL sample of serum is pulled through the cartridge, with the sedatives and matrix interferents retained following a liquid–solid extraction (Figure 7.6.11 a). Washing the cartridge with distilled water removes any interferents (Figure 7.6.11 b). Finally, we elute the sedatives using 500 μL of acetone (Figure 7.6.11 c). In comparison to a liquid–liquid extraction, a solid phase extraction has the advantage of being easier, faster, and requires less solvent.
Continuous Extractions
An extraction is possible even if the analyte has an unfavorable partition coefficient, provided that the sample’s other components have significantly smaller partition coefficients. Because the analyte’s partition coefficient is unfavorable, a single extraction will not recover all the analyte. Instead we continuously pass the extracting phase through the sample until we achieve a quantitative extraction.
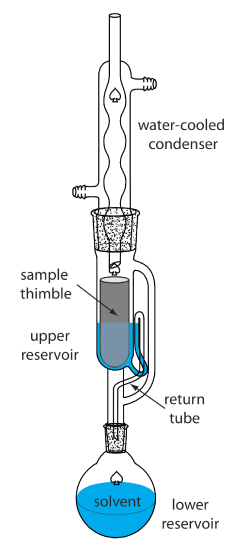
A continuous extraction of a solid sample is carried out using a Soxhlet extractor (Figure 7.6.12 ). The extracting solvent is placed in the lower reservoir and heated to its boiling point. Solvent in the vapor phase moves upward through the tube on the far right side of the apparatus, reaching the condenser where it condenses back to the liquid state. The solvent then passes through the sample, which is held in a porous cellulose filter thimble, collecting in the upper reservoir. When the solvent in the upper reservoir reaches the return tube’s upper bend, the solvent and extracted analyte are siphoned back to the lower reservoir. Over time the analyte’s concentration in the lower reservoir increases.
Microwave-assisted extractions have replaced Soxhlet extractions in some applications [Renoe, B. W. Am. Lab August 1994, 34–40]. The process is the same as that described earlier for a microwave digestion. After placing the sample and the solvent in a sealed digestion vessel, a microwave oven is used to heat the mixture. Using a sealed digestion vessel allows the extraction to take place at a higher temperature and pressure, reducing the amount of time needed for a quantitative extraction. In a Soxhlet extraction the temperature is limited by the solvent’s boiling point at atmospheric pressure. When acetone is the solvent, for example, a Soxhlet extraction is limited to 56oC, but a microwave extraction can reach 150oC.
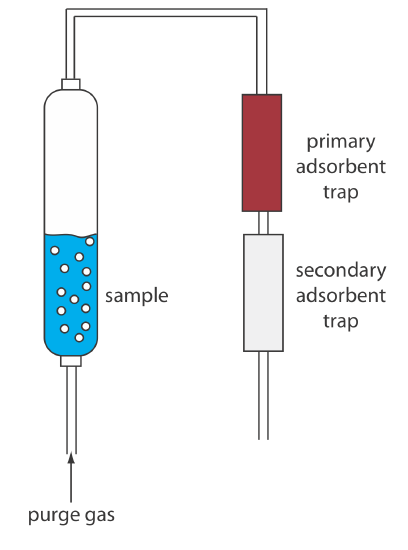
Two other continuous extractions deserve mention. Volatile organic compounds (VOCs) can be quantitatively removed from a liquid sample by a liquid–gas extraction. As shown in Figure 7.6.13 , an inert purging gas, such as He, is passed through the sample. The purge gas removes the VOCs, which are swept to a primary trap where they collect on a solid absorbent. When the extraction is complete, the VOCs are removed from the primary trap by rapidly heating the tube while flushing with He. This technique is known as a purge-and-trap. Because the analyte’s recovery may not be reproducible, an internal standard is required for quantitative work.
Continuous extractions also can be accomplished using supercritical fluids [McNally, M. E. Anal. Chem. 1995, 67, 308A–315A]. If we heat a substance above its critical temperature and pressure it forms a supercritical fluid whose properties are between those of a gas and a liquid. A supercritical fluid is a better solvent than a gas, which makes it a better reagent for extractions. In addition, a supercritical fluid’s viscosity is significantly less than that of a liquid, which makes it easier to push it through a particulate sample. One example of a supercritical fluid extraction is the determination of total petroleum hydrocarbons (TPHs) in soils, sediments, and sludges using supercritical CO2 [“TPH Extraction by SFE,” ISCO, Inc. Lincoln, NE, Revised Nov. 1992]. An approximately 3-g sample is placed in a 10-mL stainless steel cartridge and supercritical CO2 at a pressure of 340 atm and a temperature of 80oC is passed through the cartridge for 30 minutes at flow rate of 1–2 mL/min. To collect the TPHs, the effluent from the cartridge is passed through 3 mL of tetrachloroethylene at room temperature. At this temperature the CO2 reverts to the gas phase and is released to the atmosphere.
Chromatographic Separations
In an extraction, the sample originally is in one phase and we extract the analyte or the interferent into a second phase. We also can separate the analyte and interferents by continuously passing one sample-free phase, called the mobile phase, over a second sample-free phase that remains fixed or stationary. The sample is injected into the mobile phase and the sample’s components partition themselves between the mobile phase and the stationary phase. Those components with larger partition coefficients are more likely to move into the stationary phase and take longer time to pass through the system. This is the basis of all chromatographic separations. Chromatography provides both a separation of analytes and interferents, and a means for performing a qualitative or quantitative analysis for the analyte. For this reason a more thorough treatment of chromatography is found in Chapter 12.