
3.4: Selecting an Analytical Method
- Page ID
- 127190

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)A method is the application of a technique to a specific analyte in a specific matrix. We can develop an analytical method to determine the concentration of lead in drinking water using any of the techniques mentioned in the previous section. A gravimetric method, for example, might precipiate the lead as PbSO4 or as PbCrO4, and use the precipitate’s mass as the analytical signal. Lead forms several soluble complexes, which we can use to design a complexation titrimetric method. As shown in Figure 3.2.1, we can use graphite furnace atomic absorption spectroscopy to determine the concentration of lead in drinking water. Finally, lead’s multiple oxidation states (Pb0, Pb2+, Pb4+) makes feasible a variety of electrochemical methods.
Ultimately, the requirements of the analysis determine the best method. In choosing among the available methods, we give consideration to some or all the following design criteria: accuracy, precision, sensitivity, selectivity, robustness, ruggedness, scale of operation, analysis time, availability of equipment, and cost.
Accuracy
Accuracy is how closely the result of an experiment agrees with the “true” or expected result. We can express accuracy as an absolute error, e
\[e = \text{obtained result} - \text{expected result} \nonumber\]
or as a percentage relative error, %er
\[\% e_r = \frac {\text{obtained result} - \text{expected result}} {\text{expected result}} \times 100 \nonumber\]
A method’s accuracy depends on many things, including the signal’s source, the value of kA in Equation 3.3.1 or Equation 3.3.2, and the ease of handling samples without loss or contamination. A total analysis technique, such as gravimetry and titrimetry, often produce more accurate results than does a concentration technique because we can measure mass and volume with high accuracy, and because the value of kA is known exactly through stoichiometry.
Because it is unlikely that we know the true result, we use an expected or accepted result to evaluate accuracy. For example, we might use a standard reference material, which has an accepted value, to establish an analytical method’s accuracy. You will find a more detailed treatment of accuracy in Chapter 4, including a discussion of sources of errors.
Precision
When a sample is analyzed several times, the individual results vary from trial-to-trial. Precision is a measure of this variability. The closer the agreement between individual analyses, the more precise the results. For example, the results shown in the upper half of Figure 3.4.1 for the concentration of K+ in a sample of serum are more precise than those in the lower half of Figure 3.4.1 . It is important to understand that precision does not imply accuracy. That the data in the upper half of Figure 3.4.1 are more precise does not mean that the first set of results is more accurate. In fact, neither set of results may be accurate.
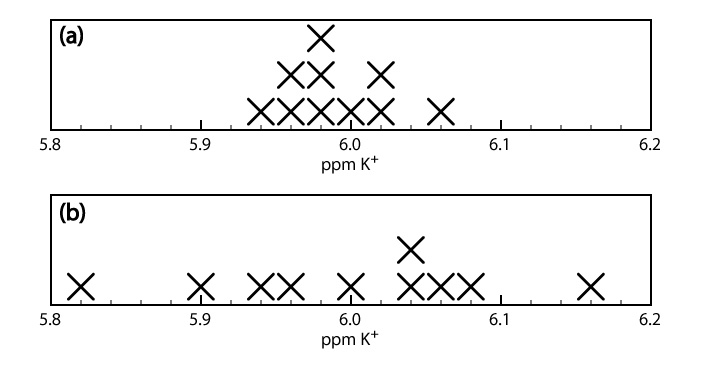
A method’s precision depends on several factors, including the uncertainty in measuring the signal and the ease of handling samples reproducibly. In most cases we can measure the signal for a total analysis technique with a higher precision than is the case for a concentration method.
Confusing accuracy and precision is a common mistake. See Ryder, J.; Clark, A. U. Chem. Ed. 2002, 6, 1–3, and Tomlinson, J.; Dyson, P. J.; Garratt, J. U. Chem. Ed. 2001, 5, 16–23 for discussions of this and other common misconceptions about the meaning of error. You will find a more detailed treatment of precision in Chapter 4, including a discussion of sources of errors.
Sensitivity
The ability to demonstrate that two samples have different amounts of analyte is an essential part of many analyses. A method’s sensitivity is a measure of its ability to establish that such a difference is significant. Sensitivity is often confused with a method’s detection limit, which is the smallest amount of analyte we can determine with confidence.
Confidence, as we will see in Chapter 4, is a statistical concept that builds on the idea of a population of results. For this reason, we will postpone our discussion of detection limits to Chapter 4. For now, the definition of a detection limit given here is sufficient.
Sensitivity is equivalent to the proportionality constant, kA, in Equation 3.3.1 and Equation 3.3.2 [IUPAC Compendium of Chemical Terminology, Electronic version]. If \(\Delta S_A\) is the smallest difference we can measure between two signals, then the smallest detectable difference in the absolute amount or the relative amount of analyte is
\[\Delta n_A = \frac {\Delta S_A} {k_A} \quad \text{ or } \quad \Delta C_A = \frac {\Delta S_A} {k_A} \nonumber\]
Suppose, for example, that our analytical signal is a measurement of mass using a balance whose smallest detectable increment is ±0.0001 g. If our method’s sensitivity is 0.200, then our method can conceivably detect a difference in mass of as little as
\[\Delta n_A = \frac {\pm 0.0001 \text{ g}} {0.200} = \pm 0.0005 \text{ g} \nonumber\]
For two methods with the same \(\Delta S_A\), the method with the greater sensitivity—that is, the method with the larger kA—is better able to discriminate between smaller amounts of analyte.
Specificity and Selectivity
An analytical method is specific if its signal depends only on the analyte [Persson, B-A; Vessman, J. Trends Anal. Chem. 1998, 17, 117–119; Persson, B-A; Vessman, J. Trends Anal. Chem. 2001, 20, 526–532]. Although specificity is the ideal, few analytical methods are free from interferences. When an interferent contributes to the signal, we expand Equation 3.3.1 and Equation 3.3.2 to include its contribution to the sample’s signal, Ssamp
\[S_{samp} = S_A + S_I = k_A n_A + k_I n_I \label{3.1}\]
\[S_{samp} = S_A + S_I = k_A C_A + k_I C_I \label{3.2}\]
where SI is the interferent’s contribution to the signal, kI is the interferent’s sensitivity, and nI and CI are the moles (or grams) and the concentration of interferent in the sample, respectively.
Selectivity is a measure of a method’s freedom from interferences [Valcárcel, M.; Gomez-Hens, A.; Rubio, S. Trends Anal. Chem. 2001, 20, 386–393]. A method’s selectivity for an interferent relative to the analyte is defined by a selectivity coefficient, KA,I
\[K_{A,I} = \frac {k_I} {k_A} \label{3.3}\]
which may be positive or negative depending on the signs of kI and kA. The selectivity coefficient is greater than +1 or less than –1 when the method is more selective for the interferent than for the analyte.
Although kA and kI usually are positive, they can be negative. For example, some analytical methods work by measuring the concentration of a species that remains after is reacts with the analyte. As the analyte’s concentration increases, the concentration of the species that produces the signal decreases, and the signal becomes smaller. If the signal in the absence of analyte is assigned a value of zero, then the subsequent signals are negative.
Determining the selectivity coefficient’s value is easy if we already know the values for kA and kI. As shown by Example 3.4.1 , we also can determine KA,I by measuring Ssamp in the presence of and in the absence of the interferent.
A method for the analysis of Ca2+ in water suffers from an interference in the presence of Zn2+. When the concentration of Ca2+ is 100 times greater than that of Zn2+, an analysis for Ca2+ has a relative error of +0.5%. What is the selectivity coefficient for this method?
Solution
Since only relative concentrations are reported, we can arbitrarily assign absolute concentrations. To make the calculations easy, we will let CCa = 100 (arbitrary units) and CZn = 1. A relative error of +0.5% means the signal in the presence of Zn2+ is 0.5% greater than the signal in the absence of Zn2+. Again, we can assign values to make the calculation easier. If the signal for Cu2+ in the absence of Zn2+ is 100 (arbitrary units), then the signal in the presence of Zn2+ is 100.5.
The value of kCa is determined using Equation 3.3.2
\[k_\text{Ca} = \frac {S_\text{Ca}} {C_\text{Ca}} = \frac {100} {100} = 1 \nonumber\]
In the presence of Zn2+ the signal is given by Equation 3.4.2; thus
\[S_{samp} = 100.5 = k_\text{Ca} C_\text{Ca} + k_\text{Zn} C_\text{Zn} = (1 \times 100) + k_\text{Zn} \times 1 \nonumber\]
Solving for kZn gives its value as 0.5. The selectivity coefficient is
\[K_\text{Ca,Zn} = \frac {k_\text{Zn}} {k_\text{Ca}} = \frac {0.5} {1} = 0.5 \nonumber\]
If you are unsure why, in the above example, the signal in the presence of zinc is 100.5, note that the percentage relative error for this problem is given by
\[\frac {\text{obtained result} - 100} {100} \times 100 = +0.5 \% \nonumber\]
Solving gives an obtained result of 100.5.
Wang and colleagues describe a fluorescence method for the analysis of Ag+ in water. When analyzing a solution that contains \(1.0 \times 10^{-9}\) M Ag+ and \(1.1 \times 10^{-7}\) M Ni2+, the fluorescence intensity (the signal) was +4.9% greater than that obtained for a sample of \(1.0 \times 10^{-9}\) M Ag+. What is KAg,Ni for this analytical method? The full citation for the data in this exercise is Wang, L.; Liang, A. N.; Chen, H.; Liu, Y.; Qian, B.; Fu, J. Anal. Chim. Acta 2008, 616, 170-176.
- Answer
-
Because the signal for Ag+ in the presence of Ni2+ is reported as a relative error, we will assign a value of 100 as the signal for \(1 \times 10^{-9}\) M Ag+. With a relative error of +4.9%, the signal for the solution of \(1 \times 10^{-9}\) M Ag+ and \(1.1 \times 10^{-7}\) M Ni2+ is 104.9. The sensitivity for Ag+ is determined using the solution that does not contain Ni2+; thus
\[k_\text{Ag} = \frac {S_\text{Ag}} {C_\text{Ag}} = \frac {100} {1 \times 10^{-9} \text{ M}} = 1.0 \times 10^{11} \text{ M}^{-1} \nonumber\]
Substituting into Equation \ref{3.2} values for kAg, Ssamp , and the concentrations of Ag+ and Ni2+
\[104.9 = (1.0 \times 10^{11} \text{ M}^{-1}) \times (1 \times 10^{-9} \text{ M}) + k_\text{Ni} \times (1.1 \times 10^{-7} \text{ M}) \nonumber\]
and solving gives kNi as \(4.5 \times 10^7\) M–1. The selectivity coefficient is
\[K_\text{Ag,Ni} = \frac {k_\text{Ni}} {k_\text{Ag}} = \frac {4.5 \times 10^7 \text{ M}^{-1}} {1.0 \times 10^{11} \text{ M}^{-1}} = 4.5 \times 10^{-4} \nonumber\]
A selectivity coefficient provides us with a useful way to evaluate an interferent’s potential effect on an analysis. Solving Equation \ref{3.3} for kI
\[k_I = K_{A,I} \times k_A \label{3.4}\]
and substituting in Equation \ref{3.1} and Equation \ref{3.2}, and simplifying gives
\[S_{samp} = k_A \{ n_A + K_{A,I} \times n_I \} \label{3.5}\]
\[S_{samp} = k_A \{ C_A + K_{A,I} \times C_I \} \label{3.6}\]
An interferent will not pose a problem as long as the term \(K_{A,I} \times n_I\) in Equation \ref{3.5} is significantly smaller than nA, or if \(K_{A,I} \times C_I\) in Equation \ref{3.6} is significantly smaller than CA.
Barnett and colleagues developed a method to determine the concentration of codeine (structure shown below) in poppy plants [Barnett, N. W.; Bowser, T. A.; Geraldi, R. D.; Smith, B. Anal. Chim. Acta 1996, 318, 309– 317]. As part of their study they evaluated the effect of several interferents. For example, the authors found that equimolar solutions of codeine and the interferent 6-methoxycodeine gave signals, respectively of 40 and 6 (arbitrary units).
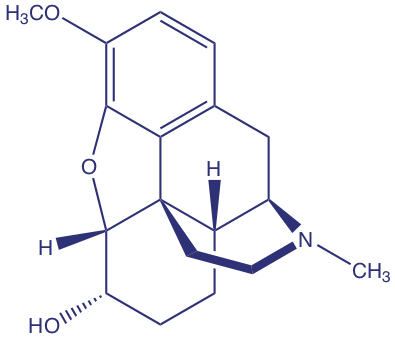
(a) What is the selectivity coefficient for the interferent, 6-methoxycodeine, relative to that for the analyte, codeine.
(b) If we need to know the concentration of codeine with an accuracy of ±0.50%, what is the maximum relative concentration of 6-methoxy-codeine that we can tolerate?
Solution
(a) The signals due to the analyte, SA, and the interferent, SI, are
\[S_A = k_A C_A \quad \quad S_I = k_I C_I \nonumber\]
Solving these equations for kA and for kI, and substituting into Equation \ref{3.4} gives
\[K_{A,I} = \frac {S_I / C_I} {S_A / C_I} \nonumber\]
Because the concentrations of analyte and interferent are equimolar (CA = CI), the selectivity coefficient is
\[K_{A,I} = \frac {S_I} {S_A} = \frac {6} {40} = 0.15 \nonumber\]
(b) To achieve an accuracy of better than ±0.50% the term \(K_{A,I} \times C_I\) in Equation \ref{3.6} must be less than 0.50% of CA; thus
\[K_{A,I} \times C_I \le 0.0050 \times C_A \nonumber\]
Solving this inequality for the ratio CI/CA and substituting in the value for KA,I from part (a) gives
\[\frac {C_I} {C_A} \le \frac {0.0050} {K_{A,I}} = \frac {0.0050} {0.15} = 0.033 \nonumber\]
Therefore, the concentration of 6-methoxycodeine must be less than 3.3% of codeine’s concentration.
When a method’s signal is the result of a chemical reaction—for example, when the signal is the mass of a precipitate—there is a good chance that the method is not very selective and that it is susceptible to an interference.
Mercury (II) also is an interferent in the fluorescence method for Ag+ developed by Wang and colleagues (see Practice Exercise 3.4.1). The selectivity coefficient, KAg,Hg has a value of \(-1.0 \times 10^{-3}\).
(a) What is the significance of the selectivity coefficient’s negative sign?
(b) Suppose you plan to use this method to analyze solutions with concentrations of Ag+ no smaller than 1.0 nM. What is the maximum concentration of Hg2+ you can tolerate if your percentage relative errors must be less than ±1.0%?
- Answer
-
(a) A negative value for KAg,Hg means that the presence of Hg2+ decreases the signal from Ag+.
(b) In this case we need to consider an error of –1%, since the effect of Hg2+ is to decrease the signal from Ag+. To achieve this error, the term \(K_{A,I} \times C_I\) in Equation \ref{3.6} must be less than –1% of CA; thus
\[K_\text{Ag,Hg} \times C_\text{Hg} = -0.01 \times C_\text{Ag} \nonumber\]
Substituting in known values for KAg,Hg and CAg, we find that the maximum concentration of Hg2+ is \(1.0 \times 10^{-8}\) M.
Problems with selectivity also are more likely when the analyte is present at a very low concentration [Rodgers, L. B. J. Chem. Educ. 1986, 63, 3–6].
Look back at Figure 1.1.1, which shows Fresenius’ analytical method for the determination of nickel in ores. The reason there are so many steps in this procedure is that precipitation reactions generally are not very selective. The method in Figure 1.1.2 includes fewer steps because dimethylglyoxime is a more selective reagent. Even so, if an ore contains palladium, additional steps are needed to prevent the palladium from interfering.
Robustness and Ruggedness
For a method to be useful it must provide reliable results. Unfortunately, methods are subject to a variety of chemical and physical interferences that contribute uncertainty to the analysis. If a method is relatively free from chemical interferences, we can use it to analyze an analyte in a wide variety of sample matrices. Such methods are considered robust.
Random variations in experimental conditions introduces uncertainty. If a method’s sensitivity, k, is too dependent on experimental conditions, such as temperature, acidity, or reaction time, then a slight change in any of these conditions may give a significantly different result. A rugged method is relatively insensitive to changes in experimental conditions.
Scale of Operation
Another way to narrow the choice of methods is to consider three potential limitations: the amount of sample available for the analysis, the expected concentration of analyte in the samples, and the minimum amount of analyte that will produce a measurable signal. Collectively, these limitations define the analytical method’s scale of operations.
We can display the scale of operations visually (Figure 3.4.2 ) by plotting the sample’s size on the x-axis and the analyte’s concentration on the y-axis. For convenience, we divide samples into macro (>0.1 g), meso (10 mg–100 mg), micro (0.1 mg–10 mg), and ultramicro (<0.1 mg) sizes, and we divide analytes into major (>1% w/w), minor (0.01% w/w–1% w/w), trace (10–7% w/w–0.01% w/w), and ultratrace (<10–7% w/w) components. Together, the analyte’s concentration and the sample’s size provide a characteristic description for an analysis. For example, in a microtrace analysis the sample weighs between 0.1 mg and 10 mg and contains a concentration of analyte between 10–7% w/w and 10–2% w/w.
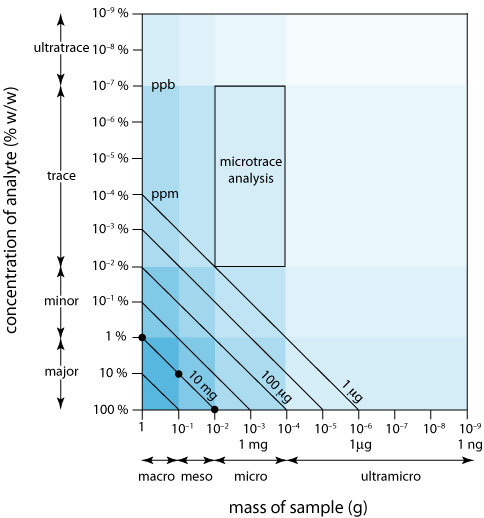
The diagonal lines connecting the axes show combinations of sample size and analyte concentration that contain the same absolute mass of analyte. As shown in Figure 3.4.2 , for example, a 1-g sample that is 1% w/w analyte has the same amount of analyte (10 mg) as a 100-mg sample that is 10% w/w analyte, or a 10-mg sample that is 100% w/w analyte.
We can use Figure 3.4.2 to establish limits for analytical methods. If a method’s minimum detectable signal is equivalent to 10 mg of analyte, then it is best suited to a major analyte in a macro or meso sample. Extending the method to an analyte with a concentration of 0.1% w/w requires a sample of 10 g, which rarely is practical due to the complications of carrying such a large amount of material through the analysis. On the other hand, a small sample that contains a trace amount of analyte places significant restrictions on an analysis. For example, a 1-mg sample that is 10–4% w/w in analyte contains just 1 ng of analyte. If we isolate the analyte in 1 mL of solution, then we need an analytical method that reliably can detect it at a concentration of 1 ng/mL.
It should not surprise you to learn that a total analysis technique typically requires a macro or a meso sample that contains a major analyte. A concentration technique is particularly useful for a minor, trace, or ultratrace analyte in a macro, meso, or micro sample.
Equipment, Time, and Cost
Finally, we can compare analytical methods with respect to their equipment needs, the time needed to complete an analysis, and the cost per sample. Methods that rely on instrumentation are equipment-intensive and may require significant operator training. For example, the graphite furnace atomic absorption spectroscopic method for determining lead in water requires a significant capital investment in the instrument and an experienced operator to obtain reliable results. Other methods, such as titrimetry, require less expensive equipment and less training.
The time to complete an analysis for one sample often is fairly similar from method-to-method. This is somewhat misleading, however, because much of this time is spent preparing samples, preparing reagents, and gathering together equipment. Once the samples, reagents, and equipment are in place, the sampling rate may differ substantially. For example, it takes just a few minutes to analyze a single sample for lead using graphite furnace atomic absorption spectroscopy, but several hours to analyze the same sample using gravimetry. This is a significant factor in selecting a method for a laboratory that handles a high volume of samples.
The cost of an analysis depends on many factors, including the cost of equipment and reagents, the cost of hiring analysts, and the number of samples that can be processed per hour. In general, methods that rely on instruments cost more per sample then other methods.
Making the Final Choice
Unfortunately, the design criteria discussed in this section are not mutually independent [Valcárcel, M.; Ríos, A. Anal. Chem. 1993, 65, 781A–787A]. Working with smaller samples or improving selectivity often comes at the expense of precision. Minimizing cost and analysis time may decrease accuracy. Selecting a method requires carefully balancing the various design criteria. Usually, the most important design criterion is accuracy, and the best method is the one that gives the most accurate result. When the need for a result is urgent, as is often the case in clinical labs, analysis time may become the critical factor.
In some cases it is the sample’s properties that determine the best method. A sample with a complex matrix, for example, may require a method with excellent selectivity to avoid interferences. Samples in which the analyte is present at a trace or ultratrace concentration usually require a concentration method. If the quantity of sample is limited, then the method must not require a large amount of sample.
Determining the concentration of lead in drinking water requires a method that can detect lead at the parts per billion concentration level. Selectivity is important because other metal ions are present at significantly higher concentrations. A method that uses graphite furnace atomic absorption spectroscopy is a common choice for determining lead in drinking water because it meets these specifications. The same method is also useful for determining lead in blood where its ability to detect low concentrations of lead using a few microliters of sample is an important consideration.