
16.16: Equilibrium Constants Revisited
- Page ID
- 49580

In discussing entropy and spontaneity, we have tended to treat chemical reactions as though they had no other alternatives than either going to completion or not occurring at all. It is only when both reactants and products are pure solids or pure liquids, however, that we find this all-or-nothing-at-all type of behavior. Reactions which involve gases or solutions are governed by an equilibrium constant, and as we saw in the sections on chemical equilibrium, this means that there is always some reactant and some product in the equilibrium mixture. Consequently such reactions must always occur to some extent, no matter how minute, and can never go quite to completion.
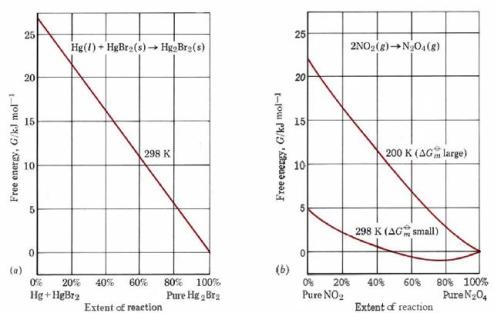
Figure \(\PageIndex{1}\) illustrates how the free energy G varies as the reaction proceeds in the two cases. If only pure solids and liquids are involved, then a plot of G against the extent of the reaction is a straight line, as shown in part a of the figure for the reaction
\[\ce{Hg(l) + HgBr2(s)→Hg2Br2(s)}\qquad 1 \text{ atm, 298K} \nonumber \]
In such a case, if ΔGm°, the free-energy difference between reactants and products, is negative, then the reaction will attain the lowest value of G possible by going to completion.
When gases and solutions are involved in the reaction, a plot of G against the extent of the reaction is no longer a straight line but exhibits a “sag,“ as shown in Figure \(\PageIndex{1}\) b for the reaction
\[\ce{2NO2(g)→N2O4(g)}\qquad 1 \text{ atm}\ \label{2} \]
at two different temperatures. In such a case, even though ΔGm° is negative, the reaction will not go to completion but will end up at the lowest point of the curve. If the value of ΔGm° is quite small, below about 10 or 20 kJ mol–1, this results in an eventual equilibrium mixture containing an appreciable proportion of both reactants and products. This is the case for Eq. \(\ref{2}\) at 298 K, when the reaction attains only 81 percent completion at equilibrium. A more usual situation is that shown for this same reaction at 200 K. Because the free-energy difference is larger at this temperature, the “sag” in the curve is much smaller, and as a result the minimum value of G lies extremely close to 100 percent completion (actually 99.9 percent). As a rough rule of thumb, therefore, we can say that if ΔGm° is negative and numerically greater than 20 kJ mol–1, the reaction will go virtually to completion; if ΔGm° is positive and larger than 20 kJ mol–1, the reaction will scarcely occur at all. Between the limits ΔGm° ± 20 kJ mol–1, we can expect measurable quantities of both reactant and product to be present at equilibrium. The above discussion suggests that there must be some relationship between the free-energy change ΔGm° and the equilibrium constant. The derivation of this relationship is too complex to produce here, so that only the result will be given. For gases the relationship has the form
\[{\Delta G_{m}^{ o}}={-RT}{\text{ ln } K^{o}} \label{3} \]
or, if base 10 logarithms are used,
\[{\Delta G_{m}^{ o}}={-2.303 RT}{\text{ ln } K^{o}} \label{4} \]
Where K° (K standard) is called the standard equilibrium constant. K° is closely related to Kp and differs from it only by being a dimensionless number. Recall from the section on the equilibrium constant in terms of pressure that Kp is defined for the general chemical reaction
\[ a \text{A} + b \text{B} + \cdots \rightleftharpoons c \text{C} + d \text{D} + \cdots \nonumber \]
by the equation
\[K_{p}=\dfrac{p_{\text{C}}^{c}\text{ }\times \text{ }p_{\text{D}}^{d}\text{ }\times \text{ }\cdots }{p_{\text{A}}^{a}\text{ }\times \text{ }p_{\text{B}}^{b}\text{ }\times \text{ }\cdots } \nonumber \]
etc., are partial pressures. The definition of K° is identical except that it involves pressure ratios (i.e., pure numbers) rather than pressures:
\[K\text{ }\!\!{}^\circ\!\!\text{ }=\dfrac{\left( \dfrac{p_{\text{C}}}{p\text{ }\!\!{}^\circ\!\!\text{ }} \right)^{c}\text{ }\times \text{ }\left( \dfrac{p_{\text{D}}}{p\text{ }\!\!{}^\circ\!\!\text{ }} \right)^{d}\text{ }\times \text{ }\cdots }{\left( \dfrac{p_{\text{A}}}{p\text{ }\!\!{}^\circ\!\!\text{ }} \right)^{a}\text{ }\times \text{ }\left( \dfrac{p_{\text{B}}}{p\text{ }\!\!{}^\circ\!\!\text{ }} \right)^{b}\text{ }\times \text{ }\cdots } \nonumber \]
Where p° is a standard pressure—almost always 1 atm (101.325 kPa). Thus if the partial pressures pa, pb, etc., are expressed in atmospheres, K° involves the same number as Kp.
The equilibrium constant Kp for the reaction
\[ \ce{2NO2 <=> N2O4} \nonumber \]
is 0.0694 kPa–1 at 298 K. Use this value to find ΔGm°(298 K) for the reaction.
Solution
We must first express Kp in atmospheres:
\[K_{p}=\text{0}\text{.0694 kPa}^{-\text{1}}=\dfrac{\text{0}\text{.0694}}{\text{kPa}}\text{ }\times \text{ }\dfrac{\text{101}\text{.3 kPa}}{\text{1 atm}}=\text{7}\text{.03 atm}^{-\text{1}} \nonumber \]
Thus
\( K^\circ = 7.03 \qquad \text{ a pure number} \)
From Eq. \(\ref{4}\) we now have
\(\begin{align} \Delta G_m^{\circ} & = –2.303RT \text{ log } K^{\circ} \\ &= – 8.3143 \text{ J K}^{-1} \text{ mol}^{-1} \times 298 \text{ K} \times 2.303 \times \text{ log } 7.03 \\ & = –4833 \text{ J mol}^{-1} = –4.833 \text{ kJ mol}^{-1} \end{align} \)
Note: You may also use ln x as well as log x as long as you use Eq. \(\ref{3}\) directly:
\[ \Delta G_m^{\circ} = –RT \text{ ln } K^{\circ} = – RT \times \text{ ln } 7.03 = – RT \times 1.950 = –4.833 \nonumber \]
In the section on the molecular view of equilibrium, we argued that the value of an equilibrium constant is the product of two factors:
\[{K}=\text{energy factor}\times\text{probability factor} \label{14} \]
The energy factor takes account of the fact that a higher-energy species is less likely to occur in an equilibrium mixture than a lower-energy species, especially at low temperatures. The probability factor reflects the fact that if there are a larger number of ways in which a molecule can arrange itself in space, a molecule is more likely to occur in that state than in one for which a smaller number of spatial arrangements is possible.
We are now in a position to make quantitative the qualitative argument presented. Combining
\[{\Delta G_{m}^{o}}={\Delta H_{m}^{ o}}-{T\Delta S_{m}^{ o}} \nonumber \]
with
\[{\Delta G_{m}^{o}}=\text{RT}{\text{ ln }{K^o}} \nonumber \]
we find
\[{-}\text{RT}{\text{ ln }{K^o}}={\Delta H_{m}^{ o}}-{T\Delta S_{m}^{ o}} \nonumber \]
giving us
\[{ \text{ ln }{K^o}}=\dfrac{-\Delta H_{m}^{ o}}{RT} + \dfrac{\Delta S_{m}^{ o}}{R} \label{18} \]
or, in base 10 logarithms
\[{ \text{ log }{K^o}}=\dfrac{-\Delta H_{m}^{ o}}{\text{2}\text{.303}RT} + \dfrac{\Delta S_{m}^{ o}}{\text{2}\text{.303}R} \label{19} \]
If we now take the logarithm of each side of Eq. \(\ref{14}\), we have
\[\text{ log }{K}=\text{ log }(\text{energy factor})+{\text{ log }(\text{probability factor})}\, \nonumber \]
This equation has the same form as Eq. \(\ref{19}\), and if we confine ourselves to the standard equilibrium constant K°, we can say that
\[ \text{ log } (\text{energy factor})=\dfrac{-\Delta H_{m}^{ o}}{\text{2}\text{.303}RT} \label{21} \]
and
\[ \text{ log } (\text{probability factor})=\dfrac{\Delta S_{m}^{ o}}{\text{2}\text{.303}R} \label{22} \]
Although we did not formally derive Eq. \(\ref{3}\), on which these results are based, we can examine the last two equalities [Eqs. \(\ref{21}\) and \(\ref{22}\)] to see that they agree with our qualitative expectations. If ΔHm° is negative, we know that the products of a reaction have a lower enthalpy (and hence lower energy) than the reactants. Thus we would predict that products would he favored by the energy factor, and this is what Eq. \(\ref{21}\) says—the more negative ΔHm°, the more positive the logarithm of the energy factor and the larger the standard equilibrium constant. Also, since T appears in the denominator of the right-hand side of Eq. \(\ref{21}\), the smaller the value of T, the larger (and hence more important) the energy factor for a given value of ΔHm°. We have also seen in the section on thermodynamic probability and entropy that an increase in entropy of a system corresponds to an increase in thermodynamic probability. This is precisely what Eq. \(\ref{22}\) says. The larger the value of ΔSm°, the larger the probability factor and hence the larger the standard equilibrium constant. Thus our qualitative description of the two factors affecting the equilibrium constant has been refined to the point where macroscopic quantities, ΔHm° and ΔSm°, can be related to what is happening on the microscopic level.
In addition to the feature we have just mentioned, Eq. \(\ref{19}\) is useful in another way. If we can measure or estimate ΔHm° and ΔSm° at temperatures other than the usual 298.15 K, we can obtain K° and calculate the extent of reaction as shown in the next example.
Find the concentration of NO in equilibrium with air at 1000 K and 1 atm pressure.
\[ \ce{N2 (g) + O2 (g) -> 2NO (g) } \Delta H_m^{\circ} = 181 \text{kJ mol}^{-1} \nonumber \]
\[ \Delta S_m^{\circ} = 35.6 \text{J K}^{-1} \text{ mol}^{-1} \nonumber \]
Solution
We first find K° :
\[{\text{ log }{K^o}}=\dfrac{-\Delta H_{m}^{ o}}{\text{2}\text{.303}RT} + \dfrac{\Delta S_{m}^{ o}}{\text{2}\text{.303}R}= \text{–9.45 + 1.34}= \text{–8.11} \nonumber \]
Thus
\[ K^{\circ} = 10^{-8.11} = 7.71 \times 10^{-9} \nonumber \]
However, since Δn = 0, \(K^{\circ} = K_p = 7.71 \times 10^{-9}\)
Assuming the air to be 80% N2 and 20% O2, we can write
\[ p_{\text{N2}} = 0.8 \text{ atm} ~~~~~\text{and}~~~~~ p_{\text{O2}} = 0.2 \text{ atm} \nonumber \]
Thus
\[{K}_{p}=\dfrac{p_{\text{NO}_{\text{2}}}^{\text{2}}}{\text{(}p_{\text{N}_{\text{2}}}\text{)(}p_{\text{O}_{\text{2}}}\text{)}} = \dfrac{p_{\text{NO}_{\text{2}}}^{\text{2}}}{\text{0}\text{.8 atm }\times \text{ 0}\text{.2 atm}} = {\text{7.71}}\times{\text{10}}^{-\text{9}} \nonumber \]
Giving
\(p_{\text{NO}_{\text{2}}}^{\text{2}} = {\text{7.71}}\times{\text{10}}^{-\text{9}}\times {\text{0.16 atm}}^{\text{2}}={\text{1.23}}\times{\text{10}}^{-\text{9}}{\text{atm}}^{\text{2}}\)
\({p}_{\text{NO}_2}=\sqrt{\text{1}\text{.23 }\times \text{ 10}^{-9}\text{ atm}^{\text{2}}}= {\text{3.5}}\times{\text{10}}^{- \text{5}}{\text{atm}} ={\text{3.5}}\times{\text{10}}^{- \text{3}}{\text{kPa}}\)
But
\({c}_{\text{NO}_2}=\dfrac{c_{\text{NO}_{\text{2}}}}{V} = \dfrac{p_{\text{NO}_{\text{2}}}}{RT}\)
Thus \[{c}_{\text{NO}_2}=\dfrac{\text{3}\text{.5 }\times \text{ 10}^{-\text{3}}\text{ kPa}}{\text{8}\text{.3143 J K}^{-\text{1}}\text{ mol}^{-\text{1}}}\text{ }\times \text{ }\dfrac{\text{1}}{\text{1000 K}} = {\text{4.3}}\times{\text{10}}^{- \text{7}}{\text{dm}}^{- \text{3}} \nonumber \]
Almost all combustion processes heat N2 and O2 to high temperatures, producing small concentrations of NO. Under most circumstances this is not of great consequence, but in the presence of sunlight and partially burned gasoline, NO2 can initiate a form of air pollution called photochemical smog. The presence of minute concentrations of NO in the upper atmosphere from high-flying supersonic jet airplanes can also deplete the ozone layer there.