
6: Classification and Catalytic Strategies of Enzymes
- Page ID
- 165295

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Classification
http://nptel.ac.in/courses/104102016/36
Enzymes are generally named according to the reaction they catalyze or by suffixing “ase” after the name of substrate. The International Union of Biochemistry and Molecular Biology developed a nomenclature for enzymes. Each enzyme is described by a sequence of four numbers preceded by "EC". EC denotes Enzyme Commission and the number of enzyme is called EC numbers.
When classified, each enzyme is assigned the EC number, in the form of digits separated by periods. The first number categorizes the enzyme based on its reaction.
- The first three numbers represent the class, subclass and sub-subclass to which an enzyme belongs, and the fourth digit is a serial number to identify the particular enzyme within a sub-subclass.
- The class, subclass and sub-subclass provide additional information about the reaction classified. For example, in the case of EC 1.2.3.4, the digits indicate that the enzyme is an oxidoreductase (class 1), that it acts on the aldehyde or oxo group of donors (subclass 2), that oxygen is an acceptor (sub-subclass 3) and that it was the fourth enzyme classified in this sub-subclass (serial number 4).
- The last printed list of enzymes appeared in the year 1992. Since then it has been updated and maintained online.
Accordingly the enzymes are classified into seven main family classes and many sub-family classes.
Oxidoreductases, Transferases, Hydrolases, Lyases, Isomerases, Ligases, Translocases
https://en.wikibooks.org/wiki/Struct...Classification
https://www.creative-enzymes.com/resource/enzyme-definition-and-classification_18.html
Catalytic Strategies
Enzymes are the most diverse type of protein in a cell. They vary not only in size, but also in the number of independently manufactured subunits that must come together to form an active enzyme, or holoenzyme. Part of the reason for requiring so many different enzymes is that they are usually very specific for their substrate molecules, and that specificity is based upon a combination of shape and charge. The interactions between substrate and enzyme are often likened to a lock and key or pieces of a jigsaw puzzle. If the substrate fits the shape of the enzyme’s active site (the part of the enzyme that carries out the actual catalytic reaction), and the charges interact (e.g. positively charged amino acids on the enzyme lining up with negative charges on the substrate), then there may be further stabilization of the interaction by Van der Waals and hydrogen bond interactions. In fact, formation of a stable Enzyme-Substrate (ES) intermediate is energetically analogous to the transition state of reactions.
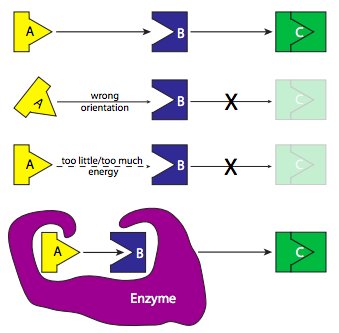
The specificity of enzymes is such that stereoisomers may not be recognized by some enzymes: for example, a protease (enzymes that chop up proteins into smaller pieces by hydrolyzing the peptide bonds between specific amino acids) such as trypsin can be stymied by the presence of a D-amino acid in place of the usual L-amino acid in a protein, even though it is a mirror image of the very same amino acid. This specificity means that enzymes are highly selective with respect to the reactions they catalyze, which means that specific reactions can be greatly enhanced without causing a general increase in many related chemical reactions. Another implication of the high specificity is that enzymes can (and often do) have high affinity for their substrates without the problem of binding non-substrate molecules.
If most biochemical reactions would proceed extremely slowly, if at all, without catalysis, enzymes are needed to lower the activation energy needed for chemical reactions to support life. Exactly how does an enzyme lower the activation energy of a reaction? What exactly does “activation energy” mean in the context of a cell? To understand this, there are two principles to keep in mind: first, when we talk about chemical reactions, generally, we are concerned with populations of substrate, product, and enzyme molecules, not individuals; and second, the reactions are generally taking place between molecules dissolved in the aqueous cytoplasm of the cell.
Once a substrate has been bound, it is the enzyme's job to quickly transform the substrate into product. The enzyme does so by carrying the substrate over a catalytic pathway. In a catalytic pathway, the reaction takes a different course than it would on its own. Sometimes the catalytic pathway is longer, involving additional steps, but because the energetic terrain is easier to traverse than in the un-catalyzed process, the catalytic reaction actually takes much less time.
Enzymes have a range of structures and reaction properties, so there are a wide number of different reactions they can catalyze. Nevertheless, there are a few common strategies displayed in catalytic reactions that are useful to know.
Approximation
When we make an approximation, we are getting close to the answer. If someone asks us what time it is, and it is 3:02 pm, we probably tell them it's about three o'clock. That's close enough.
In enzyme catalysis, approximation means getting things close to each other. If we have a substrate that is going to react with something, the enzyme can bind the substrate in such a way as to get the substrate in close proximity to the reactant. Sometimes, but not always, that might mean binding two substrates, so that they can more easily react with each other. If you want to meet your friend, it's a much better idea to say, let's meet at Cafe Santropol at 7:00, rather than wander the streets of Montreal, along with four million other people, hoping to bump into your friend. The reactant and substrate are much more likely to encounter each other in the protected confines of the enzyme than they are floating around in the wilderness of the cell.
- In approximation, two substrates are held close together within the enzyme
- This proximity makes them react together more easily
Sometimes, this idea can be a little more subtle. Imagine that the reactant is the enzyme itself, so that just by binding the single substrate, the reaction is already much more likely to occur. The substrate may be held in such a way that it is already in close proximity to amino acid side chains that will work on it and transform it into a new molecule.
- In approximation, the substrate is held in position so that subsequent reaction is much more likely
Approximation is really an entropy factor. By binding the substrate, we can limit its degrees of freedom, restricting its location or even its orientation so that there is no way the reactant can miss its intended target.
Of course, the substrates still have to find the active site of the enzyme. Sometimes, that task is aided by sticky surfaces on the enzyme; there can be groups on the surface of the enzyme that can interact with substrates, so that the substrates are less likely to drift away after a random collision with the enzyme. With its movement thus restricted, the substrate is more likely to move into the active site than drift off through the cell once more.
Although we are discussing enzymes in the cell, other kinds of catalysis make use of approximation as well. For example, transition metal catalysis often makes use of solid chunks of metal to catalyze the reactions of gaseous vapors. The surface of the metal gives the gas-phase molecules a place to bind, giving them a place to gather, rather than wandering around in three dimensions in the gas phase. Once they are together in one place, they are more likely to react with each other or with additional species on the surface of the metal.
Acid-Base Catalysis
Acid-base catalysis is a very common phenomenon. So many reactions involve the addition or removal of protons, especially the carbonyl reactions that are so prevalent in biochemical pathways, that proton donors and acceptors become key players. Acidic and basic side chains of the amino acids in the protein naturally fill these roles.
Acid-base catalysis can provide mechanistic advantages by rapidly enhancing the electrophilicity of a molecule. Any carbonyl compound is electrophilic, but if it gets protonated, the overall positive charge makes the carbonyl even more electrophilic.

Alternatively, acid-base catalysis might increase the nucleophilicity of a molecule. Any alcohol is nucleophilic, because it has lone pairs. However, if its proton is removed, it becomes even more nucleophilic, because of the overall negative charge.
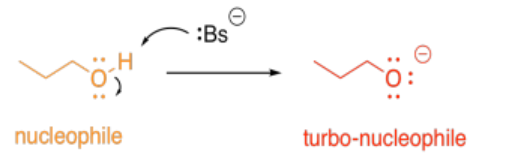
So, in the most straightforward case, adding a proton might accelerate a reaction involving an electrophile. Removing a proton might accelerate a reaction involving a nucleophile.
- Acid/base catalysis involves rapid proton shuttling
- Acidic side chains can activate electrophiles
- Basic side chains can activate nucleophiles
There are other variations on this approach. For example, consider a keto-enol tautomerism. We think of ketones classically as electrophiles, but their enol isomer is easily accessible in general, and the enol form is an excellent nucleophile. As part of a series of reaction, rapid conversion of a ketone into an enol might be a key step.
Metal Ion Catalysis
Metal ion catalysis can often be thought of as a special case of acid-base catalysis. With metal ions, we get Lewis acid catalysis. Lewis acid catalysis can accelerate reactions in a couple of different ways, in close analogy with general acid/base catalysis.
It sometimes helps to think of a metal as a great, big proton. That's an oversimplification, as we'll see in a moment. Nevertheless, it can be useful to keep the analogy in mind. When a compound binds to a metal ion, the effect can be similar to binding a proton. The compound has just donated a pair of electrons elsewhere (to the metal ion or to the proton), and so the compound suddenly looks electron-deficitient. It has enhanced electrophilicity.
On the other hand, we don't usually think of protonation as causing increased basicity; that would be completely backwards. With metals, though, that can happen, indirectly. If an alcohol, for example, donates a lone pair to a metal, the oxygen becomes positively charged. It becomes much more acidic. Suddenly, the alcohol can be deprotonated by a very, very weak base, such as water. That leads to formation of an alkoxide ion, which is much more nucleophilic than the original alcohol. Water would really never take a proton from an alcohol, but it can do it once the oxygen has a positive charge.
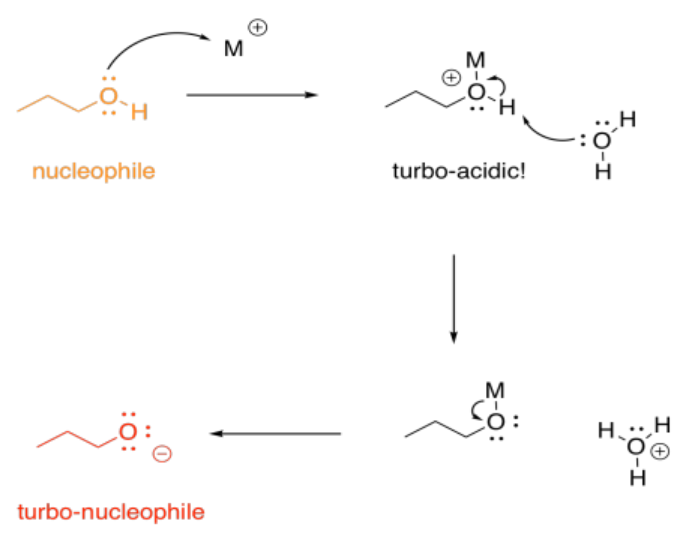
Metal ions can play a number of other roles in catalysis, but that's enough to get an idea of just some of the ways in which they might be useful. To learn more, we would have to explore more transition metal reactivity, including the ability of metals to donate and accept individual electrons.
Covalent catalysis
See 6.1 serine protease.
Suggested homework: lock-and-key model vs induced fit model.
Contributors
Contributed by E. V. Wong
Chris P Schaller, Ph.D., (College of Saint Benedict / Saint John's University)
Axolotl Academica Publishing (Biology) at Axolotl Academica Publishing