
5: Carbene Reactions
- Page ID
- 450971
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Objectives
After completing this section, you should be able to:
- Understand the structure of singlet and triplet carbenes
- Understand how to generate carbenes and carbenoids
- Understand carbene reactions involved in cyclopropantion and C-H insertion reactions
- Draw mechanisms incorporating carbenes to explain reaction outcomes
- Plan syntheses using carbene reactions
Key Terms
Make certain that you can define, and use in context, the key terms below.
- Singlet carbene
- Triplet carbene
- Carbenoid
- Alpha Elimination
- Cyclopropanation
- Simmons-Smith reaction
- C-H Insertion
Study Notes
Carbenes are the final member in the four reactive intermediates of carbon, joining carbanions, carbocations, and radicals. It is unusual to meet carbenes in Intro Orgo, so it is often a shock to realize there is a brand new version of reactive carbon that wasn't encountered earlier. Carbenes are much less important that the other three intermediates mentioned above, and we will learn only a few fundamental reactions to make them and two reactions that carbenes participate in. They provide us with one critical synthetic transformation, cyclopropanation. This reaction is the most important transformation of carbenes, and it functions as the carbon equivalent of MCPBA epoxidation. The reactivity of carbenes can be tuned by coordination with a variety of metals to form metal carbenoids. We will mention this briefly and encourage you to learn more about this fascinating research topic if you are interested.
Content
Our goals in this chapter are three-fold: 1) Understand the structure of carbenes. 2) Understand how to make carbenes and carbenoids. 3) Understand carbene cyclopropanation and carbene C-H insertion reactions. These topics cover the fundamentals of carbenes and carbene reactions and will prepare you to use and understand carbenes in the context of organic synthesis.
Carbene Structure
A carbene is a neutral form of carbon that has two bonds and two additional electrons. It is highly reactive because it has an incomplete octet with only six electrons around carbon. Carbenes can exist with their nonbonding electrons as either a lone pair or two radicals. The diradical form is known as a triplet carbene, while the structure with a lone pair is called a singlet carbene. Carbene structure is not always obvious and can depend on the method used to make the carbene. However, since we are often more comfortable thinking about carbocations and carbanions than radicals, we will simplify our analysis of carbenes and assume that they are singlet carbenes. Again, this is an oversimplification, and you are encouraged to learn more about carbenes and their structure if you are interested. Getting back to singlet carbenes, since they contain both a lone pair in a hybrid orbital (sp2) and an empty p orbital, they behave as if they are simultaneously a carbanion and a carbocation. This strange combination leads to unique reactivity. To help us recognize this type of reactivity, we will draw carbenes with both a positive and negative formal charge, thus indicating that they are neutral but highly reactive.

One relatively stable and popular class of carbenes are N-heterocyclic carbenes. These molecules are very useful as organometallic ligands, and they are seen frequently in organic synthesis.

Carbene Synthesis
The two most popular methods for the synthesis of carbenes are from alpha-elimination of a halo compound and from decomposition of a diazo compound. A common example of the alpha-elimination reaction is the deprotonation reaction of chloroform with hydroxide to yield dichlorocarbene.

Diazo decomposition is the preferred method for carbene and carbenoid generation and occurs upon exposure to heat, light, or a metal promoter (like Rh or Cu). A standard carbene can be generated from any diazo compound with heat or light with loss of nitrogen gas. Treating a diazo compound with a metal converts the diazo into a metal carbenoid. In the example below, rhodium acetate enables formation of a rhodium carbenoid. (Note: the "Ln" in the structure means some number of acetate ligands are attached to rhodium.) Rhodium and copper carbenoids have very similar reactivity to carbenes; however, the metal carbenoids are often more selective and their reactivity can be tuned based on the ligands present on the metal.

Before continuing on to carbene reactions, it is important to comment on how diazo compounds are generated. The most popular methods start with an acid chloride, an aldehyde, or a 1,3-dicarbonyl. Treating an acid chloride with two equivalents of diazomethane yields an alpha-diazo ketone. The first equivalent of diazomethane participates in a carbonyl substitution reaction. The second equivalent is the base that deprotonates the diazonium intermediate to yield the diazo product.

Treating an aldehyde with tosylhydrazine and base yields a diazo compound. As expected, tosylhydrazine reacts with the aldehyde to yield a tosylhydrazone. Methoxide removes the acidic N-H proton. The resulting anion undergoes loss of tosyl to yield the neutral diazo product.

1,3-Dicarbonyl compounds readily undergo diazo transfer reactions where an azide donates two nitrogens to form the diazo product. An example using tosyl azide is shown below. A variety of azides can participate in these reactions.

Carbene Reactions
Cyclopropanation Reactions
The most important carbene reactions are ones that enable formation of cyclopropanes. Depending on the structure of the target cyclopropane, different carbene starting materials are employed. If a dihalocyclopropane is the target, then a standard alpha-elimination reaction is used. As shown below, treating the starting alkene with chloroform and a base readily yields a dichlorocyclopropane product. The reaction mechanism is a stereospecific, concerted process.

When the goal is synthesis of a cyclopropane bearing a new CH2 group, the best option is the Simmons-Smith reaction. As shown below, combination of an alkene with diiodomethane and a Zn/Cu metallic couple yields the target cyclopropane. In the mechanism of this reaction, a free carbene is not formed. Instead, zinc first does an oxidative addition with one of the C-I bonds. The resulting organozinc intermediate behaves like a carbene, participating in a concerted reaction with the alkene to yield the cyclopropane and zinc(II) iodide.

Generating more highly substituted cyclopropanes involves the use of diazo compounds in both inter- and intramolecular reactions. Synthesis applications often take advantage of the increase in structural complexity available with intramolecular cyclopropanation reactions. An intermolecular example is shown below while an intramolecular reaction is featured in the following problem. The example problem below highlights a synthesis that proceeds via a copper carbenoid.

Propose the product of the following reaction.

- Answer
-
Copper reacts with the diazo compound to form a copper carbenoid. This reacts like a carbene to enable an intramolecular cyclopropanation reaction. In addition to the 3-membered ring, the reaction is selective for the production of the favored 6-membered ring as part of the bicyclic product.

C-H Insertion Reactions
C-H insertion reactions provide another reaction pathway for carbenes. (As we will see shortly, C-H insertion reactions adjacent to the carbene carbon to yield an alkene are a major limitation in carbene applications.) Intramolecular C-H insertion reactions provide another method for the synthesis of 5-membered rings. As we have seen previously, this results from a favorable 6-membered ring transition state. In the example below, a rhodium carbenoid forms first. There is no alkene for a potential cyclopropanation reaction, so a C-H insertion reaction occurs. The most favorable 6-membered ring transition state yields the 5-membered ring product.

Limitations of Carbene Reactions
When planning carbene reactions, there are several limitations to be aware of. First, don't forget that alpha-diazo ketones also participate in Wolff rearrangements (as we saw in Chapter 3 and mentioned in Exercise 3.3.8). The mechanism introduced in Chapter 3 did not involve a carbene. However, it is possible to draw an alternate mechanism that proceeds via a carbene. Compare the following mechanism to the one provided in the answer to Exercise 3.3.8. Both proceed to a ketene, they just get there in different ways. The preference for the Wolff rearrangement is to draw it as a carbene mechanism.

Second, carbenes with an adjacent C-H bond often undergo C-H insertions to yield an alkene before participating in cyclopropanation or C-H insertion to form a ring. This can be mitigated by some metal carbenoids, but it is always important to be wary of this undesired side reaction.
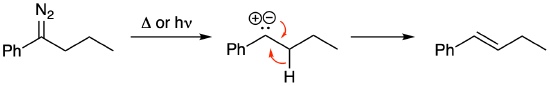
Third, nucleophilic atoms, like oxygens in ethers, can add directly to the carbene and produce rearrangement products. This is called the Stevens rearrangement and an example is provided below. The rhodium carbenoid reacts by the expected C-H insertion reaction to yield a new 6-membered ring (the ether O blocks formation of a 5-membered ring since there are no C-H bonds at this position), and it also reacts via a Stevens rearrangement to yield a 5-membered ring cyclic ether. In this mechanism, the ether O adds to the empty p orbital of the carbene then the resulting carbanion participates in a substitution reaction with the oxygen as the neutral leaving group.

Summary Problems
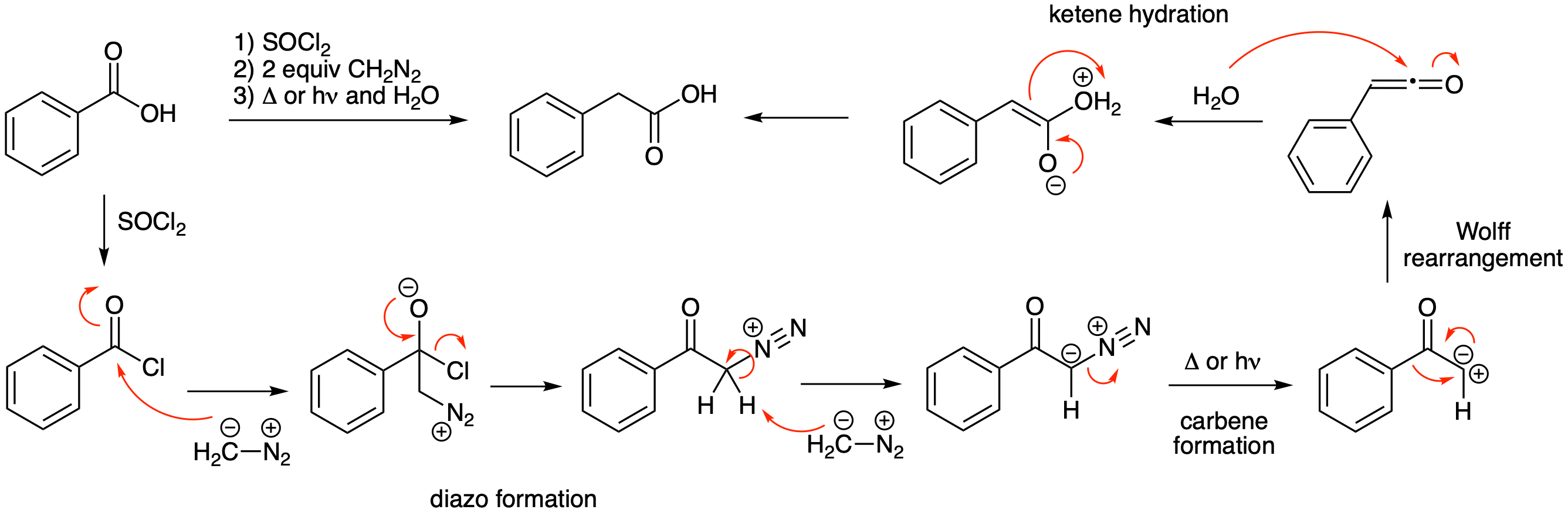
The Arndt-Eistert reaction is useful method for the homologation (extension by one carbon) of carboxylic acids. Propose a mechanism for the second and third steps in this reaction.

- Answer
-
Thionyl chloride converts the carboxylic acid into an acid chloride. As outlined previously in the chapter, conversion of an acid chloride into a diazo compound happens readily with 2 equivalents of diazomethane. Heat or light will generate the carbene that undergoes a Wolff rearrangement to yield a ketene. Hydration of the ketene yields a carboxylic acid having one more carbon than the starting material.
We mentioned the molecule bullvalene in the section on sigmatropic rearrangements in Chapter 1.4. In this problem, we will see several steps in the Doering group's synthesis of bullvalene. A key intermediate in the bullvalene synthesis is barbaralone (named for Barbara Ferrier, the first person to make it). Starting with benzene, how would you make barbaralone? Hint: One reaction in your synthesis should be the Buchner reaction.

- Answer
-
This synthesis utilizes two carbene steps. In the Buchner reaction, a carbene reacts with benzene to form a cyclopropane that quickly opens via a [3,3] sigmatropic rearrangement to yield a cycloheptatriene. This ethyl ester is hydrolyzed to the carboxylic acid and then the acid chloride. Treatment with 2 equivalents of diazomethane yields an alpha diazo ketone that reacts with copper to furnish a carbenoid that undergoes another cyclopropanation reaction to yield barbaralone.

Propose a mechanism for the following transformation.

- Answer
-
Carbenoid formation leads to an intramolecular cyclopropanation that also generates a new 5-membered ring. The cyclopropane fragments via a Cope rearrangement to form the 7-membered ring in the product. The final two steps are a base catalyzed isomerization to yield the desired target.
Literature reference - Stoltz Chemical Science 2017

Contributors
- Prof. Kevin Shea (Smith College)