
4: Radical Reactions
- Page ID
- 450970
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Objectives
After completing this section, you should be able to:
- Understand radical reactions involved in functional group conversions and carbon-carbon bond formation
- Understand radical chain reactions and radical combination reactions
- Draw mechanisms incorporating radicals to explain reaction outcomes
- Plan syntheses using radical reactions
Key Terms
Make certain that you can define, and use in context, the key terms below.
- Radical initiation
- AIBN
- Radical propagation
- Bu3SnH
- Radical termination
- Barton-McCombie deoxygenation
- Barton decarboxylation
- Hofmann-Loeffler-Freytag reaction
- Pinacol reaction
- McMurry reaction
- Acyloin reaction
Study Notes
Radical reactions are nearly always covered in Intro Orgo. Students generally learn about radical chain reactions for the bromination of alkanes (with Br2) and alkenes (with HBr and peroxides). They also often encounter allylic and benzylic bromination with NBS (a very interesting mechanism involving polar steps as part of a radical chain reaction). In this chapter, we will highlight several other useful radical chain reactions for functional group interconversions and, most importantly, for carbon-carbon bond formation. These reactions will proceed by the standard outline you have seen before: initiation with a radical initiator and propagation with a radical propagator. We will also meet synthetically useful radical reactions that proceed by a process called radical combination. These will look like termination steps from a radical chain reaction but they will be the productive pathway yielding our desired product. Radical reactions are currently a very popular research area, with many top synthetic organic labs working to develop useful transformations that are impossible using polar reactions. The information you learn in this chapter will help you understand these recent developments.
Don't forget that all mechanisms in this chapter involve the movement of single electrons (radicals!), so we will use single-headed arrows in our mechanisms.
Content
Our goal in this chapter is to introduce fundamental radical reactions that you will likely encounter in organic synthesis papers. This will build on what you have already learned in Intro Orgo about bromination reactions with bromine as the chain propagating radical by demonstrating the power of radical reactions using tributyltin radicals. These transformations will enable dehalogenation, decarboxylation, and deoxygenation reactions. Most importantly, they will demonstrate the utility of radical reactions for the synthesis of carbon-carbon bonds, especially 5- and 6-membered rings. We will also see the power of radical combination reactions for remote functionalization that can lead to the synthesis of heterocyclic ring systems.
Initiation and Propagation Steps
Before getting into the actual radical reactions, we first need to comment on two key reagents: AIBN (AzobisIsoButyroNitrile) and Bu3SnH (tributyltin hydride). We will use AIBN as our radical initiator and Bu3SnH as the precursor for our chain propagating radical, tributyltin radical. Upon heating, AIBN cleaves to form nitrogen gas and two radicals that react with Bu3SnH to yield two tributyltin radicals, as shown below. Tributyltin is our chain propagating radical, so these two radicals can each start a radical chain process (the propagation steps) resulting in the production of our desired molecule. Please remember a few key points about initiation and propagation steps: 1) We only need a small amount of the initiator to begin the chain process. Generally, 1 mol% of AIBN (0.01 equivalents) is sufficient to generate enough tributyltin radicals for the propagation steps. Although not a true catalyst, you can think about AIBN like the catalyst that must be present to make the chain reaction possible. 2) The chain propagating radical, tributyltin radical, must be a reactant in your first propagation step and a product in your last propagation step. Tributyltin radical looks a bit like a catalyst (think Pd(0) in a Stille or Suzuki reaction) but it is different in a key way. Tributyltin hydride is consumed in the reaction and must be present in a stoichiometric amount (at least 1.0 equivalents) or the chain will be broken before all of the starting material reacts. Don't forget, the propagation steps are the only productive steps in a chain reaction. This is where your product is formed. 3) Termination steps break the chain and are undesired. In radical chain reactions, combining two radicals to form a new bond is never a productive reaction. Your product will never come from a termination step. These molecules are undesired byproducts and their formation must be very limited for the radical chain reaction to be synthetically useful.

Dehalogenation Reactions
Tributyltin hydride is a useful reagent for the dehalogenation (reduction) of alkyl halides. This illustrates a radical chain mechanism using tributyltin as the chain propagating radical. (As always, AIBN and tributyltin hydride combine in the initiation steps shown above to produce tributyltin radical.) Once the tributyltin radical is formed, it reacts to form a new Sn-Br bond by cleaving the C-Br bond to generate a carbon radical. This forms tributyltin bromide, a byproduct that must be removed after the reaction, and the new carbon radical that is part of our chain process. The carbon radical reacts with tributyltin hydride (remember, we have a very small amount of AIBN, just enough to start the process, and at least 1 equivalent of Bu3SnH to react completely with the alkyl bromide starting material) to form the new C-H bond in the product and another molecule of tributyltin radical that can participate in another cycle of the chain process.

Barton-McCombie Deoxygenation
Similar to the dehalogenation reaction above, radical reactions can be used to remove an alcohol from a molecule, thus deoxygenating (reducing) the compound. We need to use some unusual chemistry to make the deoxygenation possible, specifically we need to form a xanthate, a functional group similar to a carbonate that contains two sulfur atoms. One of the sulfurs is part of a thio carbonyl (C=S) which is very reactive toward radical reactions and readily reacts with tributyltin radical to form a Sn-S bond and a carbon radical. To form the xanthate, we react the alcohol with potassium hydride to generate a negatively charged oxygen nucleophile. This reacts with carbon disulfide (the sulfur equivalent of carbon dioxide) and then methyl iodide to generate the xanthate intermediate. Once the xanthate is formed, it can react with tributyltin radical to participate in our radical chain reaction. The first step generates a new S-Sn bond and a carbon radical that decomposes to yield a dithiocarbonate ester (a byproduct that must be removed after the reaction) and a new carbon radical. At this point, the molecule has been successfully deoxygenated and the final step with tributyltin hydride completes the reaction by forming a C-H bond and regenerating the chain propagating tin radical.

Barton Decarboxylation
You likely learned about polar decarboxylation reactions in Intro Orgo. These reactions enable the removal of a carboxylic acid in a 1,3-carbonyl acid functional group. The second carbonyl positioned beta to the carboxylic acid carbonyl is critical in the mechanism (either acidic or basic conditions). These polar reactions do not work when the other carbonyl is absent. Thus, a decorboxylation reaction that doesn't require the presence of a second carbonyl at a specific position is highly valuable for synthetic chemists. This example highlights the importance of the Barton decarboxylation, a radical decarboxylation that is similar to the deoxygenation reaction shown above. It relies on a different reagent than the deoxygenation, so a xanthate ester isn't formed, but the steps are very similar, including reaction of tributyltin radical with a thiocarbonyl. In the problem below, you can propose a mechanism for this interesting reaction.
The reaction scheme below illustrates the multistep Barton decarboxylation reaction. Predict the product of the first two steps. Then, using that product, provide a mechanism for the radical decarboxylation step.

- Answer
-
The first two steps are Intro Orgo reactions. Thionyl chloride converts the carboxylic acid into an acid chloride that reacts withe the hydroxyl amine (fancy alcohol) to generate the hydroxyl amine ester product. As we saw previously, the C=S bond reacts readily with a tributyltin radical. The resulting carbon radical can generate a very stable pyridine byproduct upon cleavage of the weak N-O bond. This generates an oxygen radical that can yield carbon dioxide, thus driving this step, via C-C bond cleavage. This is the key decarboxylation step. The resulting carbon radical combines with tributyltin hydride to yield the product and regenerate the chain propagating radical.

Carbon-Carbon Bond Forming Reactions
Radical chain reactions mediated by Bu3SnH can also promote the formation of carbon-carbon bonds. These are among the most important radical reactions in synthetic chemistry since they provide us with another option in our toolbox of methods to make critical molecular connections. Key components in these reactions are a carbon-halogen bond that can react with the chain propagating tributyltin radical to generate a carbon radical that combines with an alkene or alkyne to form the new carbon-carbon bond. These reactions occur both inter- and intramolecularly with the latter being favored for higher yields and better regioselectivity. Radical reactions are kinetically controlled so they provide a very useful strategy for the synthesis of 5-membered rings. This is even true for most instances when selecting between the formation of 5- versus 6-membered rings in intramolecular cyclization reactions. (The smaller ring forms faster and, thus, is favored.) In intermolecular reactions, it is often helpful to have an electron poor alkene or alkyne as the radical partner to help favor regioselectivity with the (generally) more substituted radical formed in the first step. An example reaction and mechanism are shown below for a standard intermolecular carbon-carbon bond forming reaction. An intramolecular example is shown in the following problem.

Mechanism

For the following intramolecular C-C bond forming reaction, predict the product and propose a mechanism for its formation.

- Answer
-
Like in our intermolecular example above, the tributyltin radical attacks the carbon-halogen bond to yield a carbon radical. This new radical participates in an intramolecular C-C bond forming reaction to form a 5-membered ring radical that reacts in the final step to form the product and regenerate the chain propagating radical. Note, because of ring strain, we won't form the 4-membered ring resulting from addition to the other side of the alkene.

Predict the product of the following reaction.

- Answer
-
This problem highlights that radicals can easily form on sp2 carbons and is a reminder that bond rotation is always important to consider. The key intermediate forms and reacts to yield the new 6-membered ring which forms faster than the 7-membered ring alternative.

Non-Chain Radical Combination Reactions
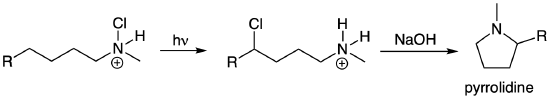
Some highly useful radical reactions involve the combination of two radicals to form an important new bond. (These look like a termination step in a radical chain mechanism.) These reactions have become increasingly relevant as new and more mild methods for Hydrogen Atom Transfer (HAT) reactions have recently been developed. We will explore some classic examples in this class of radical reactions which you can then apply to understand more contemporary transformations. The Hofmann-Loeffler-Freytag reaction enables remote functionalization of a haloamine that ultimately results in the formation of a new pyrrolidine ring (saturated 5-membered ring containing N). The reaction and mechanism are shown below. The first step in the reaction is the radical portion of the mechanism. (The chloroamine can be formed by treating the amine with t-butyl hypochlorite (tBuOCl).) Light promotes homolytic cleavage of the weak N-Cl bond to yield a Cl radical and an N radical. The next steps help explain the highly selective nature of this reaction. Bond rotation enables a HAT via a highly favored six-membered ring transition state that also transforms the reactive nitrogen radical into a more stable secondary carbon radical. The product of the first portion of the reaction forms via a radical combination reaction of the initially generated Cl radical with the newly formed C radical. This 1,4-chloroamine can undergo an intramolecular SN2 reaction to yield the pyrrolidine product. A related example of a Hoffmann-Loeffler-Freytag reaction to yield a lactam is shown in the next problem.
Mechanism

Propose a mechanism for this Hofmann-Loeffler-Freytag reaction.

- Answer
-
Iodine promotes formation of the requisite haloamine via a substitution reaction. Light initiates the radical portion of the mechanism which proceeds via the key six-membered ring transition state as shown in the prior example. After the radical combination reaction to yield the iodo amide, hydroxide deprotonates the nitrogen making the amide nucleophilic. Attack by the more reactive O yields the iminium ion-type intermediate that undergoes hydrolysis to ultimately yield the target lactone.

A related reaction promoted by lead tetraacetate (Pb(OAc)4) that proceeds via a key oxygen radical provides a synthetically useful method for the synthesis of tetrahydrofurans. As shown below, treatment of an alcohol with Pb(OAc)4 yields a substituted tetrahydrofuran (THF). The mechanism does not have a radical combination step, instead relying on a Single Electron Transfer (SET) process between a carbon radical and lead to yield a carbocation. The mechanism begins with a substitution reaction on lead with the starting alcohol. The new O-Pb bond is weak and can homolytically cleave to yield Pb(III) and an oxygen radical. Like in the Hofmann-Loefflear-Freytag reaction, the oxygen radical does a Hydrogen Atom Transfer (HAT) step via a 6-membered ring transition state to yield a carbon radical that reacts with a formally positive Pb(II) via SET. Thus, the lead is reduced and the carbon is oxidized to a carbocation that readily reacts with the intramolecular alcohol to yield the desired THF.

Mechanism

Propose a product for the following reaction.

- Answer
-
Like in the mechanism above, the lead reagent will promote formation of an oxygen radical from the alcohol. This will participate in a HAT reaction with the methyl group, followed by a SET reaction, and finally cyclization to form the new 5-membered ring.

Other useful radical combination reactions involve dimerization of carbonyl starting materials. The three most popular are the pinacol reaction, the McMurry reaction, and the acyloin reaction. These involve starting with ketones (pinacol and McMurry) or esters (acyloin) and adding a strong metal reducing agent like sodium, magnesium, or titanium to generate ketyl radicals that dimerize and ultimately produce diol (pinacol), alkene (McMurry), or keto alcohol (acyloin) products. Intermolecular examples of each are shown below. Intramolecular reactions are also possible and will occasionally show up in total synthesis papers.
Pinacol Reaction
Two equivalents of acetone react with magnesium metal via Single Electron Transfer (SET) to yield two radical anions (ketyl radical anions). These two anions react with cationic magnesium (+2) to yield the neutral diradical that undergoes a radical combination reaction to form a new carbon-carbon bond. The resulting 5-membered ring breaks down in the acidic workup to yield the diol product. In this case, the product is pinacol, the starting point for the pinacol rearrangement that we saw in Chapter 3.

McMurry Reaction
The McMurry reaction is nearly identical to the pinacol reaction except for the final step. The key difference is the use of titanium metal, generated in situ from titanium trichloride and lithium aluminum hydride. Titanium reacts with two ketones to yield two ketal radical anions that react with cationic titanium to yield a neutral diradical. The diradical participates in the radical combination reaction to yield a new 5-membered ring include a new C-C bond. At this point, the McMurry reaction diverges from the pinacol reaction. The titanocycle is not stable, instead it undergoes a deoxygenation reaction to yield an alkene product. Overall, the McMurry reaction acts like a reverse ozonolysis reaction by combining two ketones to yield an alkene.

Acyloin Reaction
The acyloin reaction begins just like the pinacol reaction with SET reactions to yield two radical anions. Dimerization (radical combination) yields the new carbon-carbon bond and an intermediate that looks like a double tetrahedral intermediate from the carbonyl addition section of Intro Orgo. Accordingly, ethoxide is a good leaving group which promotes formation of a 1,2-diketone. This compound is highly reactive toward reduction, so it will accept two more electrons from sodium to yield a diradical dianion. Radical combination generates a new pi bond. This diradical is stable and is quenched in the workup to produce a diol. Since this is an enol, it will tautomerize to yield the keto alcohol product.

Summary Problems
One of the most powerful examples of the utility of carbon-carbon bond forming radical reactions in synthesis is Curran's synthesis of hirsutene published in 1985. The reaction below is the final step in the synthesis. Propose the structure of hirsutene and a mechanism for its formation.

- Answer
-
DOI - https://doi.org/10.1021/ja00291a077
This follows the pattern we observed previously. The tin radical reacts with the iodide to form a carbon radical that reacts with the alkene to form a new 5-membered ring. The resulting tertiary radical reacts with the alkyne to form the third 5-membered ring in hirsutene. Note that the stereochemistry in the starting material determines the stereochemistry in the product. The alkyl chain coming out forms a new bond from above the initial 5-membered ring. The alkyl chain going back forms a new bond from beneath the initial 5-membered ring.

Though much less common, it is possible to use tin reagents other than tributyltin hydride to promote carbon-carbon bond forming reactions. One example is allyl tributyltin. An example of this reaction is shown below. Propose a mechanism for the initiation and propagation steps. Hint: Like in all of our other reactions with tin, tributyltin radical is the chain propagating radical.

- Answer
-
Without a Sn-H bond, radicals that react with the allyl tin reagent must add to the alkene, like we have seen in other radical reactions in this section. So, for the initiation steps, the initiating radical adds to the alkene then the resulting carbon radical decomposes to yield an allyl group on the initiating group plus the chain propagating tin radical. In the propagation steps, the first step is our standard Sn + Br to yield the first carbon radical. The only option for the new carbon radical is to add to the alkene of the tin allyl group. This generates a secondary carbon radical than decomposes just like in the initiation steps, leaving the allyl group on the product and regenerating the chain propagating tin radical. Note: alkenes are much more reactive that C-C or C-Sn single bonds.

Provide a mechanism for the reaction shown below. Hints: 1) Don't forget about the ground state structure/behavior of oxygen. 2) t-BuSH reacts similarly to Bu3SnH.

- Answer
-
The mechanism begins with standard initiation steps. AIBN generates the initiating carbon radical that reacts with t-butyl thiol to yield the chain propagating S radical. Since this behaves like the tributyltin radical, it will add to the weak C=S making a new S-S bond and carbon radical. This new radical quickly reacts to generate a stable pyridine, gaseous carbon dioxide, and a new secondary carbon radical. Remembering that oxygen exists as a diradical, we see that the next step is a radical combination between the carbon radical and O2. In the final step, the remaining O radical reacts with the beginning thiol to generate the product and regenerate the chain propagating radical.
Literature Reference: Barton Tetrahedron 1985

Provide a mechanism for the following transformation. Remember, SmI2 is an excellent single electron donor. Hint: At some point in the mechanism you will need to form an alpha-N radical (a radical on a carbon next to the N).

- Answer
-
Samarium diiodide starts the reaction by donating an electron (Single Electron Transfer (SET)), to the carbonyl. This yields a radical anion that undergoes a radical cyclization reaction to generate the new 5-membered ring and a tertiary radical. In the next step, this radical adds to the adjacent pi bond to form a transient 3-membered ring and a secondary radical. Regeneration of the alkene enables radical cleavage of the unstable cyclopropane to yield a stabilized alpha-N radical. Another SET mediated by samarium converts the radical into a carbanion. Both anions are quenched in the acidic workup to yield the target molecule.
Literature Reference: Wood Chemical Science 2020

Contributors
- Prof. Kevin Shea (Smith College)