
10.7: Additions involving cyclic intermediates
- Page ID
- 225848
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)10.7.1. Reaction of alkenes with carbenes – cyclopropanation
The highly strained nature of cyclopropane compounds makes them very reactive and interesting synthetic targets. Additionally cyclopropanes are present in many biological compounds. One common method of cyclopropane synthesis is the reaction of carbenes with the double bond in alkenes or cycloalkenes. Methylene, H2C, is the simplest carbene, and in general carbenes have the formula R2C. Other species that will also react with alkenes to form cyclopropanes but do not follow the formula of carbenes are referred to as carbenoids.
Introduction
Carbenes were once only thought of as short lived intermediates. The reactions of this section only deal with these short lived carbenes which are mostly prepared in situ, at the time of the main reaction. However, there do exist so called persistent carbenes, which are stabilized by a variety of methods often including aromatic rings or transition metals. In general a carbene is neutral and has six valence electrons, two of which are non bonding. These electrons can either occupy the same sp2 hybridized orbital to form a singlet carbene (with paired electrons), or two different sp2 orbitals to form a triplet carbene (with unpaired electrons), but we will focus exclusively on the more common singlet carbenes.
The reactivity of a singlet carbene is concerted and similar to that of electrophilic or nucleophilic addition. The highly reactive nature of carbenes leads to very fast reactions in which the rate determining step is generally carbene formation.
Preparation of methylene (:CH2)
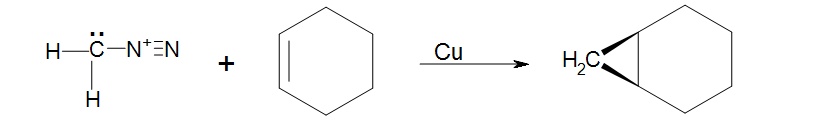
The preparation of methylene starts with the yellow gas diazomethane, CH2N2. Diazomethane can be exposed to light, heat or copper to facilitate the loss of nitrogen gas and the formation of the simplest carbene methylene. The process is driven by the formation of the nitrogen gas which is a very stable molecule.
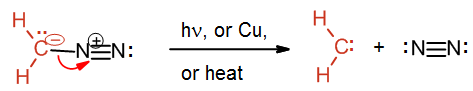
Carbene reaction with alkenes
A carbene such as methylene will react with an alkene which will break the double bond and result with a cyclopropane. The reaction will usually leave stereochemistry of the double bond unchanged. As stated before, carbenes are generally formed during the reaction; hence the starting material is diazomethane not methylene.
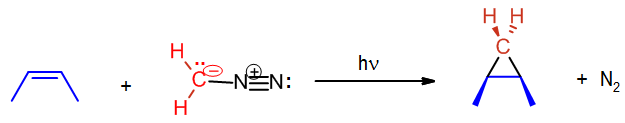
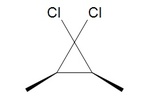
In the above case cis-2-butene is converted to cis-1,2-dimethylcyclopropane. Likewise, below the trans configuration is maintained.
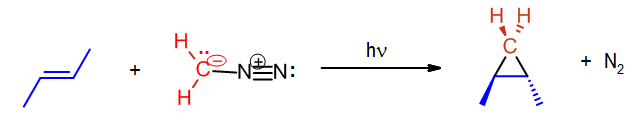
Other carbenes
In addition to the general carbene with formula R2C: there exist a number of other compounds that behave in much the same way as carbenes in the synthesis of cyclopropane derivatives. Halogenatedcarbenes such as dichlorocarbene, Cl2C:, are more stable than simple alkyl carbenes. Dichlorocarbene can be conveniently prepared from chloroform (CHCl3) with base in the presence of a phase transfer catalyst:
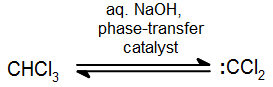
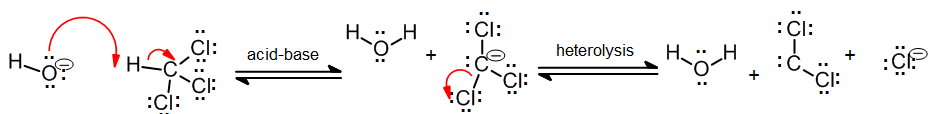
These halogenated carbenes form cyclopropanes in the same manner as methylene:
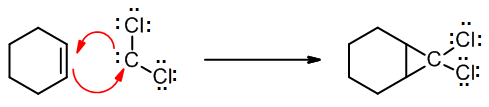
Outside links
Problems
1. Knowing that cycloalkenes react much the same as regular alkenes what would be the expected structure of the product of cyclohexene and diazomethane facilitated by copper metal?
2. What would be the result of a reaction of diazomethane with trans-3-pentene in the presence of light?
3. What starting material could be used to form cis-1,2-diethylcyclopropane?
4. What would the following reaction yield?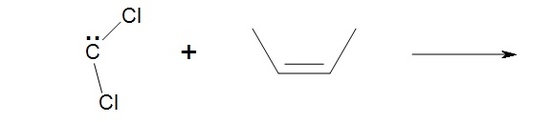
Answers
1. The product will be a bicyclic ring, Bicyclo[4.1.0]heptane.
2. The stereochemistry will be retained making a cyclopropane with trans methyl and ethyl groups. Trans-1-ethyl-2-methylcyclopropane
3. The cis configuration will be maintained from reagent to product so we would want to start with cis-3-hexene. A Simmons Smith reagent, or methylene could be used as the carbene or carbenoid.
4. The halogenated carbene will react the same as methylene yielding, cis-1,1-dichloro-2,3dimethylcyclopropane.
References
- Vollhardt, K. Peter C. and Schore, Neil E. Organic Chemistry: Structure and Function. New York: Bleyer, Brennan, 2007.
- Abdel-Wahab, Aboel-Magd A. Ahmed, Saleh A. and Dürr, Heinz. “Carbene Formation by Extrusion of Nitrogen” in CRC Handbook of Organic Photochemistry and Photobiology. CRC Press, 2004.
- Karty, J. Organic Chemistry: Principles and Mechanisms, First Edition. W. W. Norton, 2014.
10.7.2. Addition of halogens (Cl2 or Br2) to alkenes
Halogen molecules such as Cl2 and Br2 also add to alkene double bonds. Here we need not be concerned with orientation since the two ends of the adding molecule are identical, but the electrophilic addition mechanism helps us understand another characteristic of this reaction, its stereochemistry.
If bromine is added to cyclopentene, we might anticipate two products which differ in how the bromine atoms are geometrically related to each other. If the two bromines are on the same face of the ring, the compound is called cis. If they are on opposite faces, the compound is called trans. The experimental result is that only the trans product is formed.
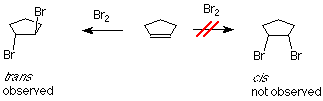
One of the bromine atoms is acting as an electrophile. If we apply the usual mechanism, its first step it would go like this:
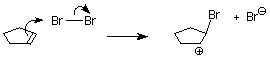
As we learned in our study of SN1 reactions, carbocations are attacked by nucleophiles on both faces. If a carbocation is present in this system, we’d expect to find both the cis and trans products.
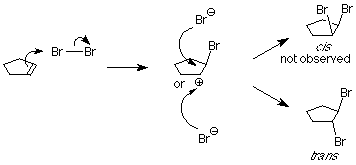
This is not what happens when the experiment is done, so we conclude that the carbocation is not present, or it reacts immediately to form a new cation. Something is happening which prevents the nucleophilic bromide ion from attacking the face of the carbocation which already is attached to the first bromine. We understand that by envisioning that the electrophilic bromine atom attaches itself to both alkene carbon atoms. One bond is made using the electrons from the pi bond, and the other is made using an unshared electron pair from the bromine. This results in a new ring formed from the bromine and the two alkene carbon atoms. It is called a “bromonium” ion. Note that the mechanism for forming the three-membered ring has some parallels with cyclopropanation, described above.
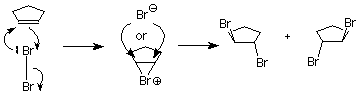
Attack by the nucleophilic bromide ion on the bromine in this ring would only result in cyclopentene and bromine, so no reaction would occur. Attack by the bromide ion on the either of the alkene carbons would be like an SN2 reaction. The attacked carbon would invert, and the product would have the trans configuration. (Notice that there are two such products, which are enantiomers, so we get a racemic mixture.)
Khan Academy on alkene halogenation:

A YouTube element has been excluded from this version of the text. You can view it online here: http://pb.libretexts.org/ochem2walker/?p=34
10.7.3. Oxymercuration-demercuration
Introduction
Carbocation rearrangement is a process in which the carbocation intermediate can form a more stable ion. With carbocation rearrangement, the reaction would not be able to hydrate quickly under mild conditions and be produced in high yields. This reaction is very fast and proceeds with 90% yield.
This reaction involves a mercury acting as a reagent attacking the alkene double bond to form a mercurinium ion Bridge. A water molecule will then attack the most substituted carbon to open the mercurium ion bridge, followed by proton transfer to solvent water molecule.
The organomercury intermediate is then reduced by sodium borohydride – the mechanism for this final step is beyond the scope of our discussion here. Notice that overall, the oxymercuration – demercuration mechanism follows Markovnikov’s regioselectivity with the OH group is attached to the more substituted carbon and the H is attach to the less substituted carbon. The reaction is useful, however, because strong acids are not required, and carbocation rearrangements are avoided because no discreet carbocation intermediate forms. However, it does require the use of highly toxic mercury compounds.
References
- Vollhardt, K. Peter C. Organic chemistry structure and function. New York: W.H. Freeman, 2007.
- Smith, Michael B., and Jerry March. March’s Advanced Organic Chemistry Reactions, Mechanisms, and Structure (March’s Advanced Organic Chemistry). New York: Wiley-Interscience, 2007 2007.
- Roderic P. Quirk , Robert E. Lea, Reductive demercuration of hex-5-enyl-1-mercuric bromide by metal hydrides. Rearrangement, isotope effects, and mechanism, J. Am. Chem. Soc., 1976, 98 (19), pp 5973–5978.
Some Practice Problems
What are the end products of these reactants?
Answers
The end product to these practice problems are pretty much very similar. First, you locate where the double bond is on the reactant side. Then, you look at what substituents are attached to each side of the double bond and add the OH group to the more substituent side and the hydrogen on the less substituent side.
Questions
Q8.4.1
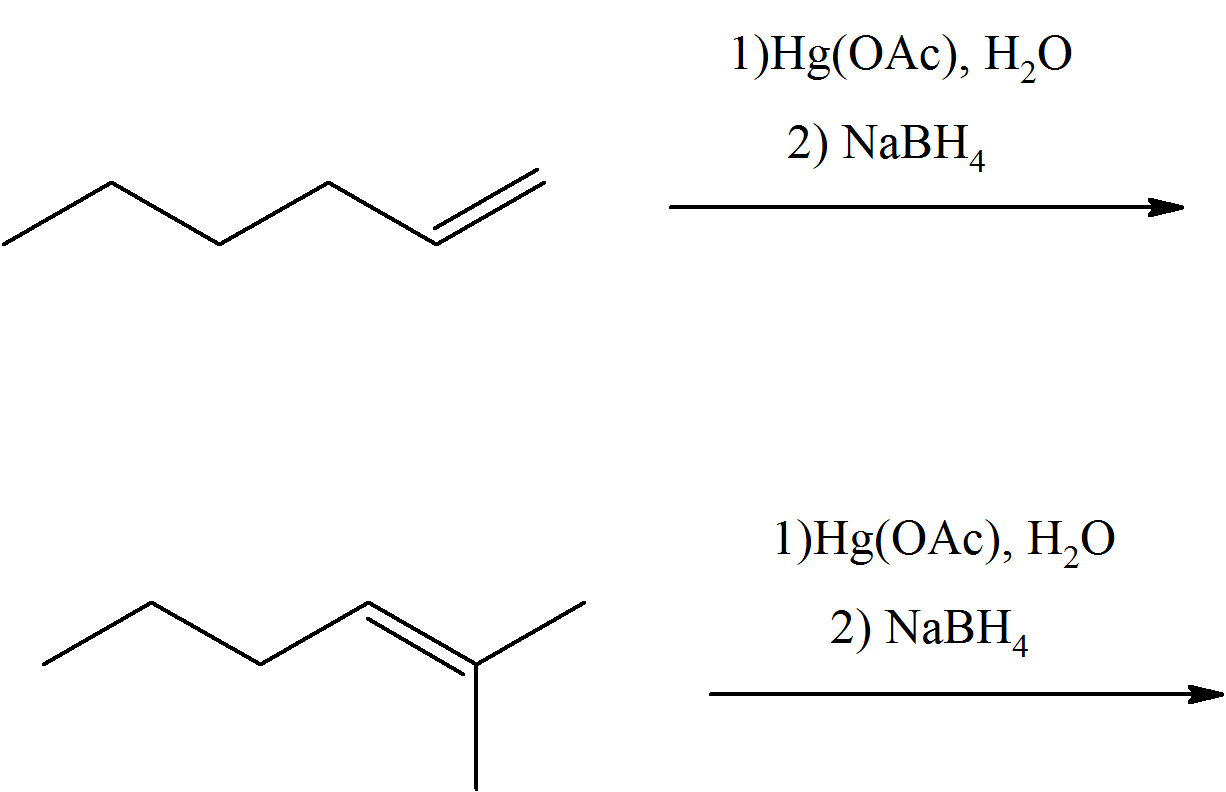
In each case, predict the product(s) of these reactants of oxymercuration.
Q8.4.2
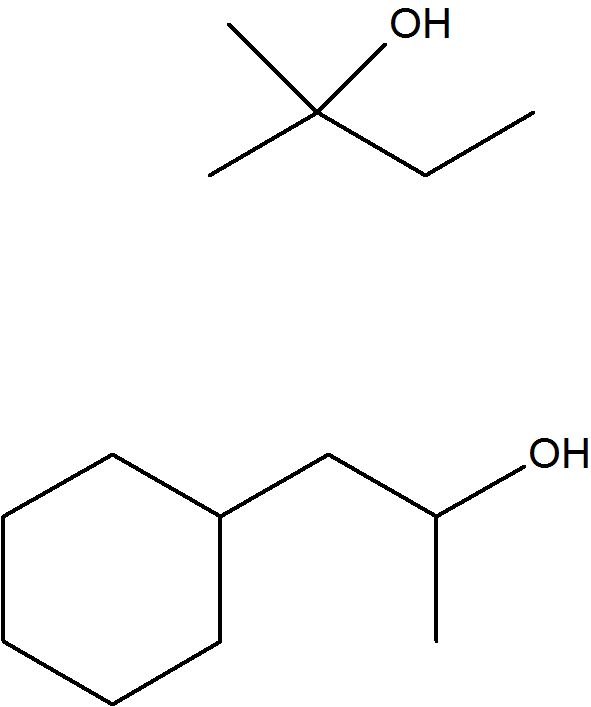
Propose the alkene that was the reactant for each of these products of oxymercuration.
Solutions
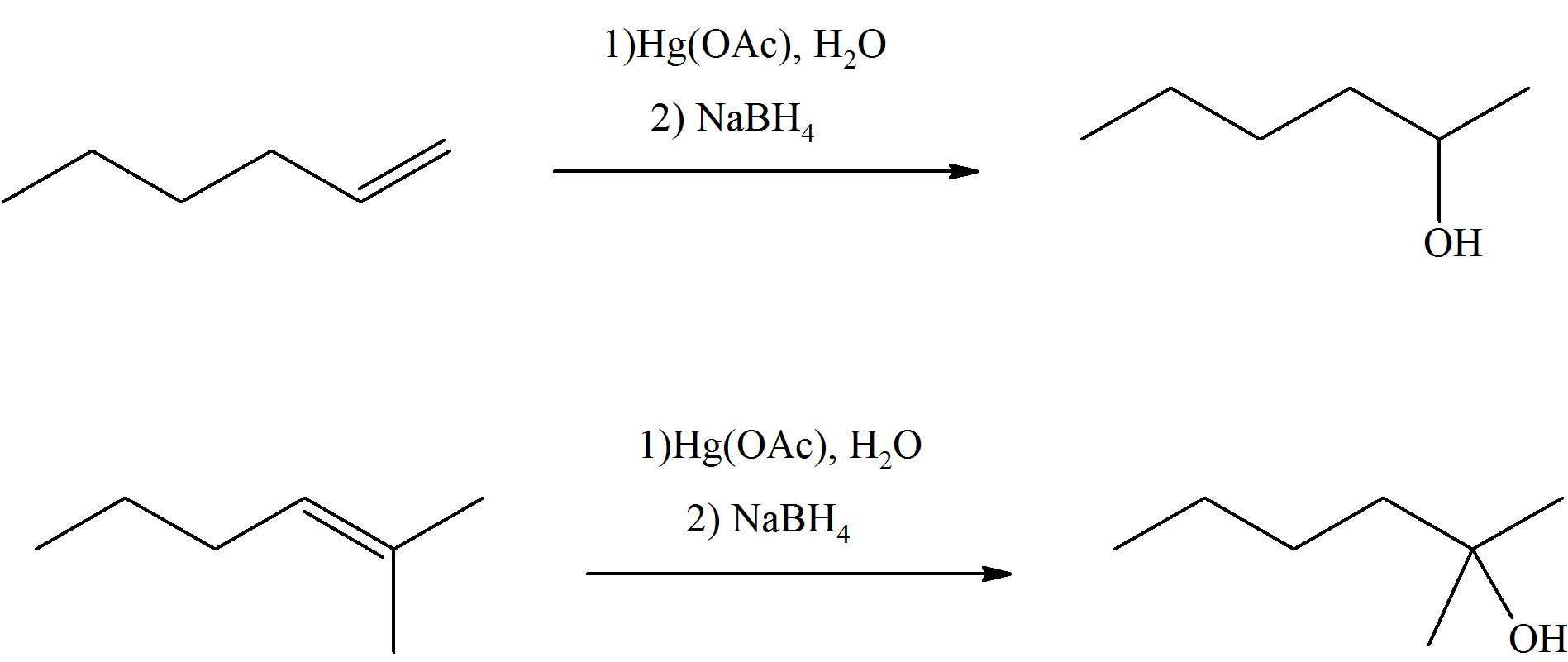
S8.4.1
S8.4.2
Video
MasterOrganicChemistry (video)

A YouTube element has been excluded from this version of the text. You can view it online here: http://pb.libretexts.org/ochem2walker/?p=34
Further Reading
- Wikipedia: Oxymercuration Reaction
- Carey 5th Ed Online: Oxymercuration-Demercuration
- Leah4Sci: Oxymercuration-Demercuration
10.7.4. Epoxide formation
Epoxide, also called oxiranes, are useful reagents that may be opened by further reaction to form anti vicinal diols. One way to synthesize epoxides is through the reaction of an alkene with a peroxycarboxylic acid such as MCPBA.
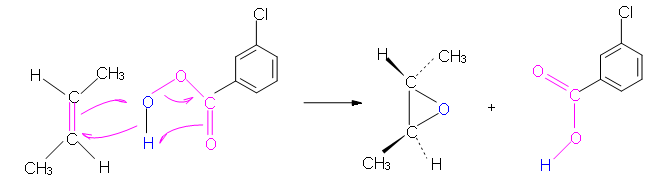
The peroxycarboxylic acid has an unusual property of having an electrophilic oxygen atom on the COOH group. The reaction is initiated by the electrophilic oxygen atom reacting with the nucleophilic carbon-carbon double bond. The mechanism involves a concerted reaction with a four-part, circular transition state. The result is that the originally electrophilic oxygen atom ends up in the epoxide ring and the C(O)OOH group becomes C(O)OH.
Mechanism
Peroxycarboxylic acids are generally unstable. One stable example is meta-chloroperoxybenzoic acid, shown in the mechanism below. Often abbreviated MCPBA, it is a stable crystalline solid that is popular for laboratory use. However, MCPBA can be explosive under some conditions.
Peroxycarboxylic acids are sometimes replaced in industrial applications by monoperphthalic acid, or the monoperoxyphthalate ion bound to magnesium, which gives magnesium monoperoxyphthalate (MMPP). In either case, a nonaqueous solvent such as dichloromethane, ether, acetone, or dioxane is used. This is because in an aqueous medium with any strong acid or base catalyst present, the epoxide ring is hydrolyzed to form a vicinal diol, a molecule with two OH groups on neighboring carbons. (For more explanation of how this reaction leads to vicinal diols, see below.) However, in a nonaqueous solvent, the hydrolysis is prevented and the epoxide ring can be isolated as the product. Reaction yields from this reaction are usually good. The reaction rate is affected by the nature of the alkene, with more nucleophilic double bonds resulting in faster reactions.
Example
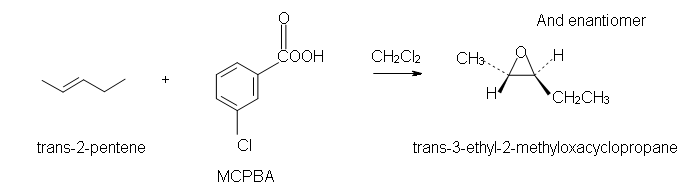 Since the transfer of oxygen is to the same side of the double bond, the resulting oxacyclopropane ring will have the same stereochemistry as the starting alkene. A good way to think of this is that the alkene is rotated so that some constituents are coming forward and some are behind. Then, the oxygen is inserted on top. (See the product of the above reaction.) One way the epoxide ring can be opened is by an acid catalyzed oxidation-hydrolysis. Oxidation-hydrolysis gives a vicinal diol, a molecule with OH groups on neighboring carbons. For this reaction, the dihydroxylation is anti since, due to steric hindrance, the ring is attacked from the side opposite the existing oxygen atom. Thus, if the starting alkene is trans, the resulting vicinal diol will have one S and one R stereocenter. But, if the starting alkene is cis, the resulting vicinal diol will have a racemic mixture of S, S and R, R enantiomers. |
References
- Royals, E. 1954. Advanced Organic Chemistry. New York: Prentice Hall. 948 p.
- Streitwieser, A. and C. Heathcock. 1981. Introduction to Organic Chemistry. 2nd ed. New York: Macmillan Publishing Co. 1258 p.
- Vollhardt, K. and N. Schore. 2007. Organic Chemistry: Structure and Function. 5th ed. New York: W.H. Freeman and Company. 1254 p.
- Wheland, G. 1949. Advanced Organic Chemistry. 3rd ed. New York: John Wiley & Sons. 871 p.
Problems
1. Predict the product of the reaction of cis-2-hexene with MCPBA (meta-chloroperoxybenzoic acid)
a) in acetone solvent.
b) in an aqueous medium with acid or base catalyst present.
2. Predict the product of the reaction of trans-2-pentene with magnesium monoperoxyphthalate (MMPP) in a chloroform solvent.
3. Predict the product of the reaction of trans-3-hexene with MCPBA in ether solvent.
4. Predict the reaction of propene with MCPBA.
a) in acetone solvent
b) after aqueous work-up.
5. Predict the reaction of cis-2-butene in chloroform solvent.
Answers
1. a) Cis-2-methyl-3-propyloxacyclopropane
b) Racemic (2R,3R)-2,3-hexanediol and (2S,3S)-2,3-hexanediol
2. Trans-3-ethyl-2-methyloxacyclopropane.
3. Trans-3,4-diethyloxacyclopropane.
4. a) 1-ethyl-oxacyclopropane
b) Racemic (2S)-1,2-propandiol and (2R)-1,2-propanediol
5. Cis-2,3-dimethyloxacyclopropane
Video

A YouTube element has been excluded from this version of the text. You can view it online here: http://pb.libretexts.org/ochem2walker/?p=34
Further Reading
- Carey 5th Ed Online: Epoxidation of Alkenes
- Leah4Sci: Alkene Epoxidation
- Chemtube3D: Oxidation of alkenes to form epoxides
10.7.5. Hydroboration-oxidation
Learning Objectives
After completing this section, you should be able to
- identify hydroboration (followed by oxidation) as a method for bringing about the (apparently) non-Markovnikov addition of water to an alkene.
- Write an equation for the formation of a trialkylborane from an alkene and borane.
- Erite an equation for the oxidation of a trialkylborane to an alcohol.
- Draw the structure of the alcohol produced by the hydroboration, and subsequent oxidation, of a given alkene.
- Determine whether a given alcohol should be prepared by oxymercuration-demercuration or by hydroboration-oxidation, and identify the alkene and reagents required to carry out such a synthesis.
- Write the detailed mechanism for the addition of borane to an alkene, and explain the stereochemistry and regiochemistry of the reaction.
Key Terms
Make certain that you can define, and use in context, the key term below.
- hydroboration
Study Notes
The two most important factors influencing organic reactions are polar (or electronic) effects and steric effects.
Hydroboration-oxidation is a two step pathway used to produce alcohols. The reaction proceeds in an anti-Markovnikov manner, where the hydrogen (from BH3 or BHR2) attaches to the more substituted carbon and the boron attaches to the least substituted carbon in the alkene double bond. Furthermore, the borane acts as a Lewis acid by accepting two electrons in its empty p orbital from an alkene that is electron rich. This process allows boron to have an complete octet. The hydroboration mechanism has the elements of both hydrogenation and electrophilic addition and it is a stereospecific (syn addition), meaning that the hydroboration takes place on the same face of the double bond, this leads cis stereochemistry.
Introduction
Hydroboration-oxidation of alkenes has been a very valuable laboratory method for the stereoselective and regioselective addition of alkenes, without any rearrangement.
The borane complex
Borane itself exists naturally as a very toxic gas in the form of a dimer with the general formula B2H6 (diborane), which ignites spontaneously in air. Borane is commercially available in ether and tetrahydrofuran (THF). In these solutions the borane exists as a Lewis acid-base complex, which allows boron to have an complete octet.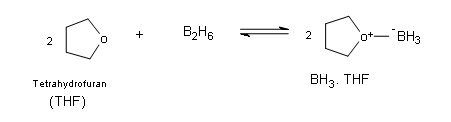
The mechanism
Elementary step #1
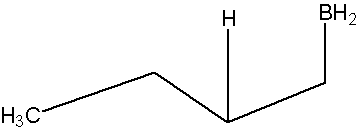
- Part #1: Hydroboration of the alkene. In this first step the addition of the borane to the alkene is initiated and proceeds as a concerted reaction because bond breaking and bond formation occur at the same time. This step involves the vacant 2p orbital of the boron electrophile pairing with the electron pair of the π-bond of the alkene nucleophile.
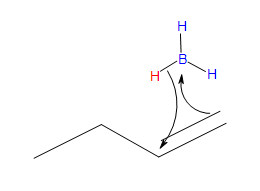
Transition state
.bmp?revision=1)
* Note that a carbocation is not formed. Therefore, no rearrangement takes place.
- In the product, the boron has added to the less substituted carbon of the alkene, which then places the hydrogen on the more substituted carbon. Both, the boron and the hydrogen add simultaneously on the same face of the double bond (syn addition).
Oxidation of the trialkylborane by hydrogen peroxide
You will not be required to know this part of the mechanism for this course, but further information is available in the links below.
Stereochemistry of hydroboration
The hydroboration reaction is among the few simple addition reactions that proceed cleanly in a syn fashion. As noted above, this is a single-step reaction. Since the bonding of the double bond carbons to boron and hydrogen is concerted, it follows that the geometry of this addition must be syn. Furthermore, rearrangements are unlikely inasmuch as a discrete carbocation intermediate is never formed. These features are illustrated for the hydroboration of α-pinene.
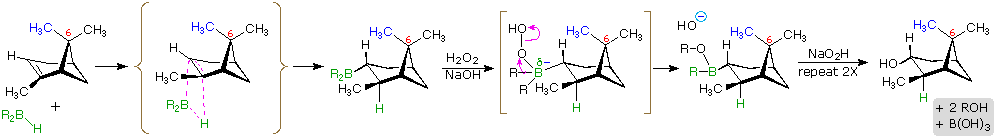
Since the hydroboration procedure is most commonly used to hydrate alkenes in an anti-Markovnikov fashion, we also need to know the stereoselectivity of the second oxidation reaction, which substitutes a hydroxyl group for the boron atom. Independent study has shown this reaction takes place with retention of configuration so the overall addition of water is also syn.
The hydroboration of α-pinene also provides a nice example of steric control in a chemical reaction. In the less complex alkenes used in earlier examples the plane of the double bond was often a plane of symmetry, and addition reagents could approach with equal ease from either side. In this case, one of the methyl groups bonded to C-6 (colored blue in the equation) covers one face of the double bond, blocking any approach from that side. All reagents that add to this double bond must therefore approach from the side opposite this methyl.
References
- Vollhardt, Peter, and Neil Shore. Organic Chemistry: Structure and Function. 5th. New York: W.H. Freeman and Company, 2007.
- Foote, S. Christopher, and William H. Brown. Organic Chemistry. 5th. Belmont, CA: Brooks/Cole Cengage Learning, 2005.
- Bruice, Paula Yurkanis. Oragnic Chemistry. 5th. CA. Prentice Hall, 2006.
- Bergbreiter E. David , and David P. Rainville. Stereochemistry of hydroboration-oxidation of terminal alkenes. J. Org. Chem., 1976, 41 (18), pp 3031–3033
- Ilich, Predrag-Peter; Rickertsen, Lucas S., and Becker Erienne. Polar Addition to C=C Group: Why Is Anti-Markovnikov Hydroboration-Oxidation of Alkenes Not “Anti-“? Journal of Chemical Education., 2006, v83, n11, pg 1681-1685
Problems
What are the products of these following reactions?
#1.
#2.
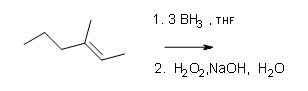
#3.
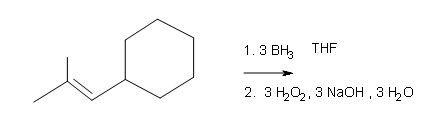
Draw the structural formulas for the alcohols that result from hydroboration-oxidation of the alkenes shown.
#4.
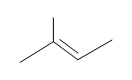
#5. (E)-3-methyl-2-pentene
If you need clarification or a reminder on the nomenclature of alkenes refer to the link below on naming the alkenes.
Answers
[reveal-answer q=”433977″]Show Solution[/reveal-answer]
[hidden-answer a=”433977″]
#1.
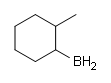
#2.
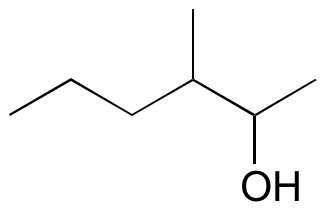
#3.
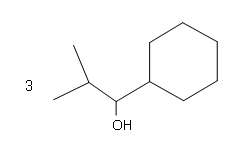
#4.
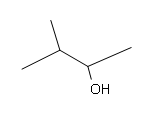
#5.
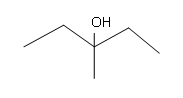
[/hidden-answer]
Exercises
Write out the reagents or products (A–D) shown in the following reaction schemes.
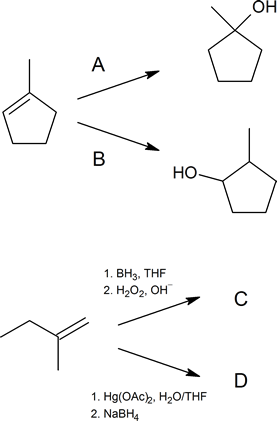
Solutions
[reveal-answer q=”652262″]Show Solution[/reveal-answer]
[hidden-answer a=”652262″]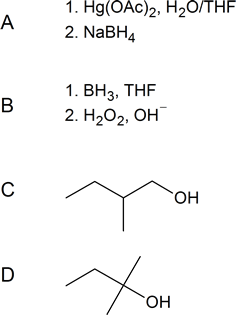 [/hidden-answer]
[/hidden-answer]
- 11.3.5 Cyclopropanation of Alkenes. Authored by: Paul Tisher. Located at: https://chem.libretexts.org/LibreTexts/Purdue/Purdue%3A_Chem_26605%3A_Organic_Chemistry_II_(Lipton)/Chapter_11.__Addition_to_pi_Systems/11.3%3A_Concerted_Additions/11.3.5_Cyclopropanation_of_Alkenes. Project: Chemistry LibreTexts. License: CC BY-NC-SA: Attribution-NonCommercial-ShareAlike
- 27: Electrophilic Additions. Authored by: Kirk McMichael (Washington State University). Located at: https://chem.libretexts.org/Textbook_Maps/Organic_Chemistry/Book%3A_Organic_Chemistry_-_A_%22Carbonyl_Early%22_Approach_(McMichael)/27%3A_Electrophilic_Additions#Halogen_Addition. Project: Chemistry LibreTexts. License: CC BY-NC-SA: Attribution-NonCommercial-ShareAlike
- 11.1.2.3: Oxymercuration. Authored by: Dr. Dietmar Kennepohl FCIC (Professor of Chemistry, Athabasca University) Lance Peery (UCD), Duyen Dao-Tran Organic Chemistry With a Biological Emphasis by Tim Soderberg (University of Minnesota, Morris) Jim Clark (Chemguide.co.uk) Prof. Steven Farmer (Sonoma State University). Located at: https://chem.libretexts.org/LibreTexts/Purdue/Purdue%3A_Chem_26605%3A_Organic_Chemistry_II_(Lipton)/Chapter_11.__Addition_to_pi_Systems/11.1%3A_Electrophilic_Addition/11.1.2_Electrophilic_Addition_to_Alkenes/11.1.2.3%3A_Oxymercuration. Project: Chemistry LibreTexts. License: CC BY-NC-SA: Attribution-NonCommercial-ShareAlike
- 11.3.6 Epoxidation of Alkenes. Authored by: Kristen Perano . Located at: https://chem.libretexts.org/LibreTexts/Purdue/Purdue%3A_Chem_26605%3A_Organic_Chemistry_II_(Lipton)/Chapter_11.__Addition_to_pi_Systems/11.3%3A_Concerted_Additions/11.3.6_Epoxidation_of_Alkenes. Project: Chemistry LibreTexts. License: CC BY-NC-SA: Attribution-NonCommercial-ShareAlike
- 9:4 Hydroboration-Oxidation: A Stereospecific Anti-Markovnikov Hydration. Authored by: Dr. Dietmar Kennepohl FCIC (Professor of Chemistry, Athabasca University) Prof. Steven Farmer (Sonoma State University) Organic Chemistry With a Biological Emphasis by Tim Soderberg (University of Minnesota, Morris) Jim Clark (Chemguide.co.uk) William Reusch, Professor Emeritus (Michigan State U.), Virtual Textbook of Organic Chemistry. Located at: https://chem.libretexts.org/LibreTexts/Winona_State_University/Klein_and_Straumanis_Guided/9%3A_Addition_Reactions_of_Alkenes/9%3A4_Hydroboration-Oxidation%3A_A_Stereospecific_Anti-Markovnikov_Hydration. Project: Chemistry LibreTexts. License: CC BY-NC-SA: Attribution-NonCommercial-ShareAlike