
1.27: Electrophilic Additions
- Page ID
- 81793

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)In the functional group of an alkene - the carbon-carbon double bond -- the most readily available electrons are those in the pi bond. They are farther from the nuclei than the electrons in a sigma bond, so they are more readily attracted to an electrophile if one approaches. Another way to say this is that the pi electrons are weakly nucleophilic. We'll begin by looking at a few reactions which begin with the attack of an electrophile on the pi part of the double bond.
First, let's briefly review a reaction of the carbonyl group with an electrophile. The acid catalyzed addition of water to an aldehyde is one such reaction discussed earlier. The mechanism is:
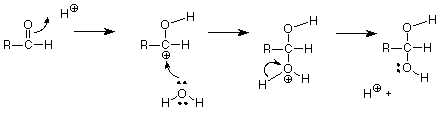
The first step is electrophilic attack on the carbonyl pi bond by the electrophilic, acid H+. This step makes a carbocation, which is then attacked by the weak but very abundant nucleophile water. (Do not use OH- as the nucleophile. There is very little OH- in an acidic solution!) The final "mop-up" step gives the product and returns an H+ to regenerate the catalyst.
Now let's apply this same mechanism to the addition of water to ethylene, the smallest alkene.
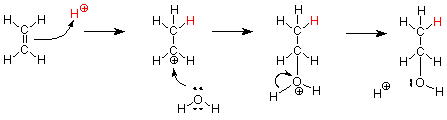
Notice that the steps are the same and that the last two steps (what happens to the carbocation) are the same steps which occured in the SN1 hydrolysis (solvolysis in water) of an alkyl halide.
These steps -- first an electrophile attacks the pi bond to form a carbocation, second a nucleophile attacks the carbocation -- are the key steps in the most important reactions of alkenes, electrophilic addition reactions. The first step is the slow one, so it is the one which determines the rate of the reaction. This has very important consequences when we introduce a slight complication into the structure of the alkene.
Orientation - Markovnikov's Rule
What happens if we apply this reaction to an alkene like propene? (Such an alkene is called "unsymmetrical" since the substitution patterns on the two alkene carbons are different.) There are two possible products since the OH could wind up on either the CH2 carbon or the CH3CH carbon.
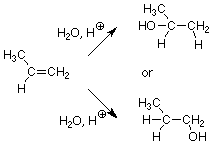
We may reasonably expect that the product which is formed fastest will be the one which predominates in the product mixture. In fact, if one product is formed more than 100 times faster than the other, it is the only product we will observe in a practical sense. This means that predicting which product is formed comes down to predicting which product is formed faster. In turn, this question becomes "which product is formed by a pathway with a lower activation energy?" We estimate relative activation energies by looking at transition state structures. The pathway with the lower energy transition state will be the faster pathway. It will produce more product in a given time than the slower pathway, so its product will be found in the greater amount.
How do we make such a prediction? We know the mechanism, so we can apply it to the pathways which would lead to our potential products. Fortunately, the rates associated with these two pathways are determined by their first steps, so we need only concern ourselves with the activation energies for those steps.
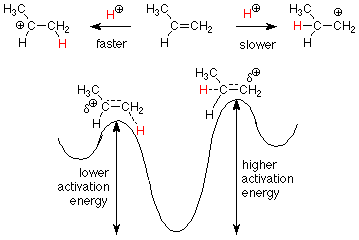
We know that a secondary carbocation (on the left) is more stable (has a lower energy) than a primary carbocation (on the right). We have explained this by saying that the the electrons in a methyl group are better at partially relieving the carbocation's electron deficiency than the relative lack of electrons around a hydrogen. Since the transition states for these two steps are also electron deficient partially formed carbocations, we would expect the same effects to prevail in the transition states. This tells us that the reaction to the left will have the lower activation energy and will occur faster. We then expect to see the product in which the attacking H+ is attached to the less substituted carbon and the OH is attached to the more substituted carbon.
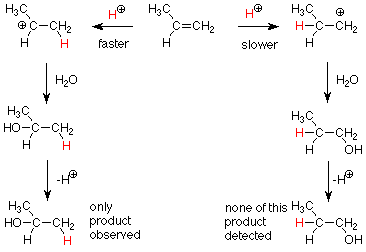
More generally, the electrophile attacks the less substituted carbon atom in the first step and the nucleophile attacks the more substituted product in the second step.
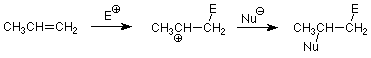
We can use this to predict which product will be formed in an electrophilic addition to an unsymmetrical alkene. All we have to do is identify the electrophile and the nucleophile in the compound which is adding. For example consider the addition of HBr. The HBr bond is polarized so that the H is positive and the Br is negative. The H+ is thus the electrophile and the Br- is the nucleophile. Application of the pattern above gives us:
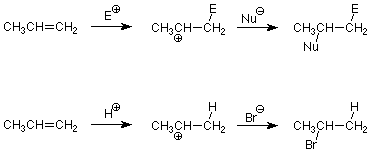
Historically, this pattern was observed by Vladimir Markovnikov in 1870, long before the mechanism was understood. The generalization that hydrogen adds to the carbon with the most hydrogens (another way to say what is in bold above) is known as Markovnikov's rule. Common applications include the additions of HBr and water (discussed above) and HCl.
Hydroboration-Oxidation
Notice that we cannot make a primary alcohol by adding water to any other alkene than ethylene by an electrophilic addition reaction as outlined above. An alternative reaction sequence which does achieve this result was developed about 40 years ago by H. C. Brown. It is outlined as follows.
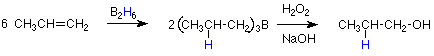
We will focus our attention on the first step. We know the product of this step, so we can identify the atoms which are acting as the electrophile and the nucleophile. The boron adds to the least substituted carbon, so it is acting as the electrophile. The hydrogen adds to the most substituted carbon, so it is acting as the nucleophile. Does this make sense? We have seen the B-H bond act this way before in the reduction of ketones and aldehydes by BH4- (in sodium borohydride). There the hydrogen acted as a nucleophile and we explained that by saying that hydrogen is more electronegative than boron. We can understand the reaction of boron as an electrophile if we consider that B2H6 dissociates into two molecules of BH3. A quick check of the electronic configuration of BH3 shows that the boron has an empty 2p orbital, so BH3 has the structural characteristics needed to qualify it as an electrophile.
Practically, the overall result of this reaction sequence is to add water to an alkene double bond in an orientation which is opposite to that predicted by Markovnikov's rule. For that reason it is referred to as an "anti-Markovnikov" addition. It is a powerful method for making primary alcohols and other alcohols where it is desired to locate the OH group on the less substituted carbon atom.
Halogen Addition
Halogen molecules such as Cl2 and Br2 also add to alkene double bonds. Here we need not be concerned with orientation since the two ends of the adding molecule are identical, but the electrophilic addition mechanism helps us understand another characteristic of this reaction, its stereochemistry.
If bromine is added to cyclopentene, we might anticipate two products which differ in how the bromine atoms are geometrically related to each other. If the two bromines are on the same face of the ring, the compound is called cis. If they are on opposite faces, the compound is called trans. The experimental result is that only the trans product is formed.
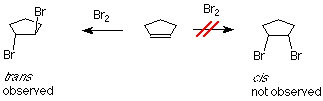
One of the bromine atoms is acting as an electrophile. If we apply the usual mechanism, it's first step it would go like this:
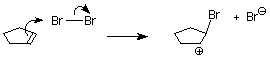
As we learned in our study of SN1 reactions, carbocations are attacked by nucleophiles on both faces. If a carbocation is present in this system, we'd expect to find both the cis and trans products.
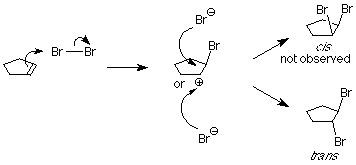
This is not what happens when the experiment is done, so we conclude that the carbocation is not present. Something else is happening, something which prevents the nucleophilic bromide ion from attacking the face of the carbocation which already is attached to the first bromine. We understand that by envisioning that the electrophilic bromine atom attaches itself to both alkene carbon atoms. One bond is made using the electrons from the pi bond, and the other is made using an unshared electron pair from the bromine. This results in a new ring formed from the bromine and the two alkene carbon atoms. It is called a "bromonium" ion.
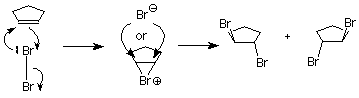
Attack by the nucleophilic bromide ion on the bromine in this ring would only result in cyclopentene and bromine, so no reaction would occur. Attack by the bromide ion on the either of the alkene carbons would be like an SN2 reaction. The attacked carbon would invert, and the product would have the trans configuration. (Notice that there are two such products, which are enantiomers, so we get a racemic mixture.)
Hydrogenation
There is another reaction of alkenes, hydrogenation, which deserves mention but which is not related to the electrophilic addition mechanism. Hydrogenation is the addition of molecular hydrogen (H22) to the alkene double bond. This converts a simple alkene into an alkane.
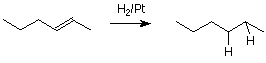
Hydrogenation reactions are carried out in the presence of a solid catalyst such as finely divided platinum (Pt) metal. The reaction occurs on the surface of the metal and involves temporary bonding of both the alkene and the hydrogen molecule to the atoms on the metal surface.
Hydrogenation is important in the conversion of highly unsaturated (many double bonds) fats to fats with fewer double bonds. This is done so that the product will have a higher melting point than the reactant and be more convenient as a dietary spread for bread. Also, the products are less susceptible to reaction with oxygen and do not turn rancid as rapidly.