
7.8: Alkene Synthesis by Dehydration of Alcohols
- Page ID
- 182934
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Learning Objective
- interpret and draw reaction energy diagrams for alcohol dehydration reactions
- propose mechanisms for dehydration reactions
- predict the products and specify the reagents for alkene synthesis from alcohol dehydration reactions
- predict and explain the stereochemistry of E2 eliminations to form alkenes, especially from cyclohexanes
Dehydration of Alcohols to Yield Alkenes
One way to synthesize alkenes is by dehydration of alcohols. Alcohols undergo E1 or E2 mechanisms to lose water and form a double bond. This mechanism is analogous to the alkyl halide mechanism. The only difference is that hydroxide is a very poor leaving group so an extra step is required. The hydroxyl group is protonated so that water is now the leaving group. Therefore, the dehydration reaction of alcohols to generate alkene proceeds by heating the alcohols in the presence of a strong acid, such as sulfuric or phosphoric acid, at high temperatures.
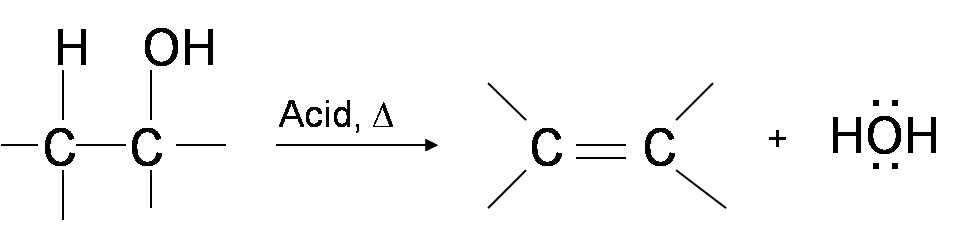
The required reaction temperature range decreases with increasing substitution of the hydroxy-containing carbon:
- 1° alcohols: 170° - 180°C
- 2° alcohols: 100°– 140 °C
- 3° alcohols: 25°– 80°C
If the reaction is not sufficiently heated, the alcohols do not dehydrate to form alkenes, but react with one another to form ethers (e.g., the Williamson Ether Synthesis).
Mechanism for the Dehydration of Alcohols
Different types of alcohols may dehydrate through a slightly different mechanism pathway. However, the general idea behind each dehydration reaction is that the –OH group in the alcohol donates two electrons to H+ from the acid reagent to form an alkyloxonium ion. This ion acts as a very good leaving group for either the E1 or E2 mechanism. The deprotonated acid (the conjugate base) then reacts with one of the beta-hydrogens to form a double bond.
Primary Alcohols and the E2 Mechanism
Primary alcohols undergo bimolecular elimination (E2 mechanism) while secondary and tertiary alcohols undergo unimolecular elimination (E1 mechanism). The relative reactivity of alcohols in dehydration reaction is ranked as the following
Methanol < primary < secondary < tertiary
Primary alcohol dehydrates through the E2 mechanism. Oxygen donates two electrons to a proton from sulfuric acid H2SO4, forming an alkyloxonium ion. The resulting conjugate base (HSO4–) approaches in an anti-coplanar orientation relative to the leaving group and reacts with one adjacent hydrogen while the alkyloxonium ion simultaneously leaves in a concerted process, making a double bond.

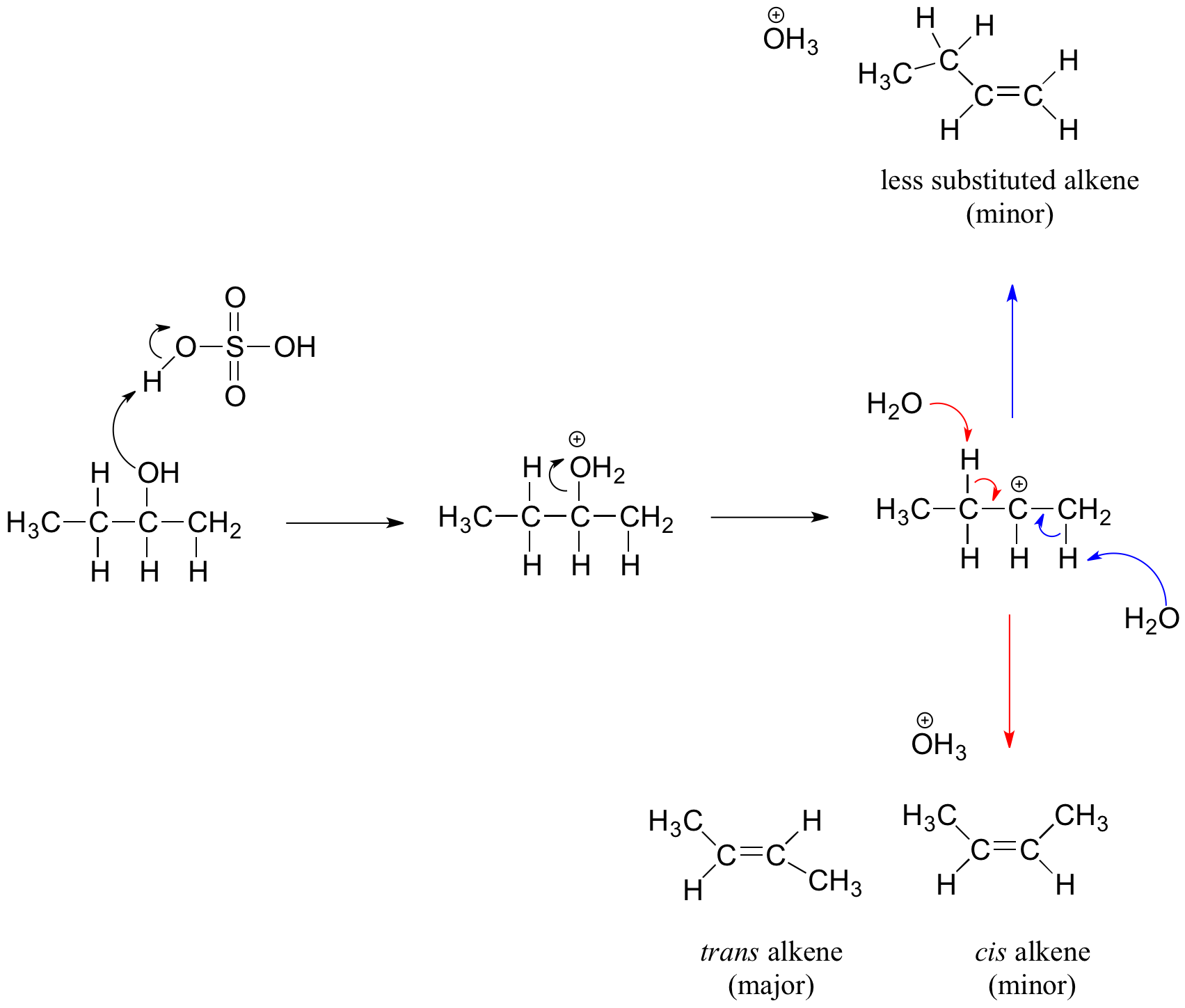
Secondary and Tertiary Alcohols and the E1 Mechanism
Secondary and tertiary alcohols dehydrate through the E1 mechanism. Similarly to the reaction above, secondary and tertiary –OH protonate to form alkyloxonium ions. However, the ion leaves first and forms a carbocation as the reaction intermediate. The water molecule (which is a stronger base than the HSO4- ion) then abstracts a proton from an adjacent carbon, forming a double bond.
For example, in the mechanism below that the alkene formed depends on which proton is abstracted: the red arrows show formation of the more substituted 2-butene, while the blue arrows show formation of the less substituted 1-butene. Recall the general rule that more substituted alkenes are more stable than less substituted alkenes, and trans alkenes are more stable than cis alkenes. Thereore, the trans diastereomer of the 2-butene product is most abundant.
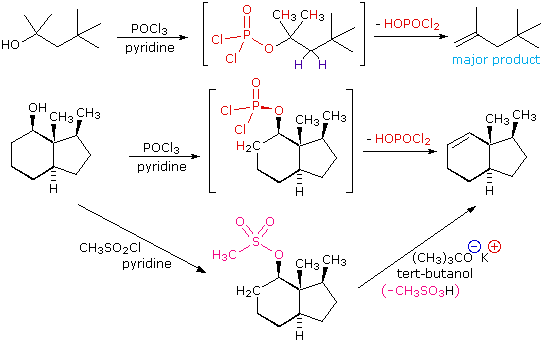
The dehydration mechanism for a tertiary alcohol is analogous to that shown above for a secondary alcohol. The E2 elimination of 3º-alcohols under relatively non-acidic conditions may be accomplished by treatment with phosphorous oxychloride (POCl3) in pyridine. This procedure is also effective with hindered 2º-alcohols, but for unhindered and 1º-alcohols an SN2 chloride ion substitution of the chlorophosphate intermediate competes with elimination. Examples of these and related reactions are given in the following figure. The first equation shows the dehydration of a 3º-alcohol. The predominance of the non-Zaitsev product (less substituted double bond) is presumed due to steric hindrance of the methylene group hydrogen atoms, which interferes with the approach of base at that site. The second example shows two elimination procedures applied to the same 2º-alcohol. The first uses the single step POCl3 method, which works well in this case because SN2 substitution is retarded by steric hindrance. The second method is another example in which an intermediate sulfonate ester confers halogen-like reactivity on an alcohol. In every case the anionic leaving group is the conjugate base of a strong acid.
Carbocation Rearrangements and the E1 Mechanism
Carbocation stability is always a driving force in E1 mechanisms. It is important to evaluate the structure of all carbocation intermediates to look for the possibility of 1,2-hydride or 1,2-methyl shifts to form more stable carbocation intermediates. Carbocation rearrangements are discussed more completely in chapter 7.
Exercise
1. Draw the bond-line structure(s) for the product(s) formed and specify the mechanism (E1 or E2) for each reaction below.
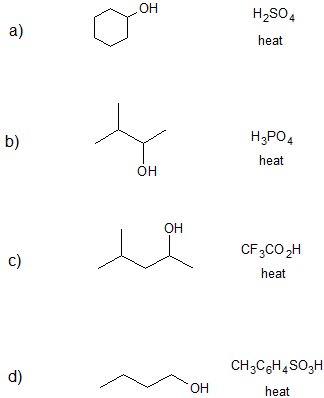
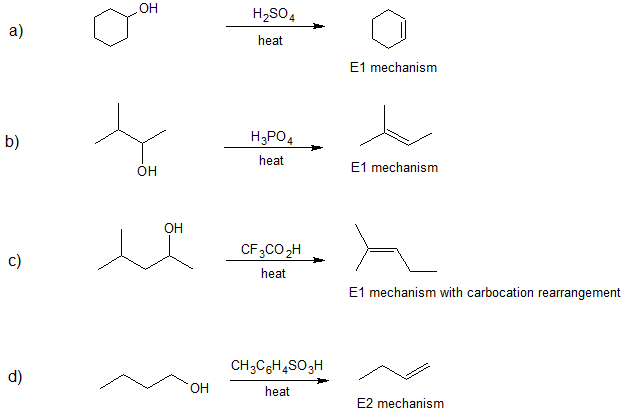
- Answer
-
Contributors and Attributions
Dr. Dietmar Kennepohl FCIC (Professor of Chemistry, Athabasca University)
Prof. Steven Farmer (Sonoma State University)
William Reusch, Professor Emeritus (Michigan State U.), Virtual Textbook of Organic Chemistry