
2.7.4: Expanded Octets and Molecular Orbitals
- Page ID
- 299375
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)In organic chemistry the octet rule is closer to a law; do not make more than four bonds to carbon! However, once you start dealing with atoms in the third row of the periodic table, the octet rule becomes a bit more of a guideline. Consider the common anion, hexfluorophospate, PF6- (Figure \(\PageIndex{1}\)), in which we draw the phosphorous atom with six bonds giving it an apparently electron count of 12.
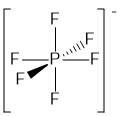
Figure \(\PageIndex{1}\). The structure of the hexafluorophosphate anion.
The bonding in these types of systems is a bit more involved and to fully appreciate it we must consider the molecular orbital diagram for such a molecule. This will be done using the same techniques as were used in the previous section, but some adjustments will need to be made as we progress from the Lewis structure to the molecular orbital diagram.
Step 1 - Drawing the structure and determining the hybridization of the central atom
Exercise \(\PageIndex{1}\)
Using VSEPR, draw the three-dimensional structure of sulfur tetrafluoride (SF4).
- Answer
-
Sulfur has 6 valence electrons and fluorine has 7 giving a total of 34 electrons in this molecule.
To start, you would draw bonds between sulfur and each of the fluorine atoms which would use 8 of the 34 electrons.
As fluorine is more electronegative, the next step would be to give each fluorine 3 lone pair in order to fill their octets. This will use 26 of the remaining electrons. At this point you might be tempted to stop as the sulfur atom and all of the fluorine atoms have an octet.
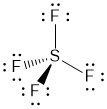
The problem is that this structure only accounts for 32 of our 34 electrons. As fluorine is an element in the 2nd row, we will not add the missing electrons to any of the fluorine atoms. That leaves sulfur as the only option. Checking the formal charge on each of the atoms supports this placement.
Formal Charge = number of valence electrons - (2 x number of lone pair + number of bonds)
FCF = 7 - (2(3) + 1) = 0
FCS = 6 - (2(1) + 4) = 0
This makes the sulfur AX4E1 so the structure is going to be based on AX5E0 which is trigonal bipyramidal.
There are two possible options for the structure, but the rules for VSEPR say that the lone pair should be as far away from other electrons as possible. In this structure that would be one of the equatorial positions and so the structure on the right (see-saw) is the correct one.
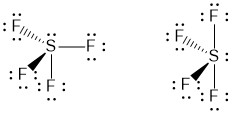
Exercise \(\PageIndex{2}\)
What orbitals need to be considered for hybridization on the central sulfur atom?
- Answer
-
We need a total of 5 hybrid orbitals to account for the 4 fluorine atoms and the lone pair. That means we need to incorporate a d-orbital which will give sp3d hybridization. The particular d-orbital that is used would be the dz2 orbital.
Step 2 - Determine the point group of the molecule and assign the generator functions
Exercise \(\PageIndex{3}\)
What is the point group of sulfur hexafluoride?
- Answer
-
SF4 does not have an infinite rotation axis.
There one rotation axis passing through the lone pair which is C2.
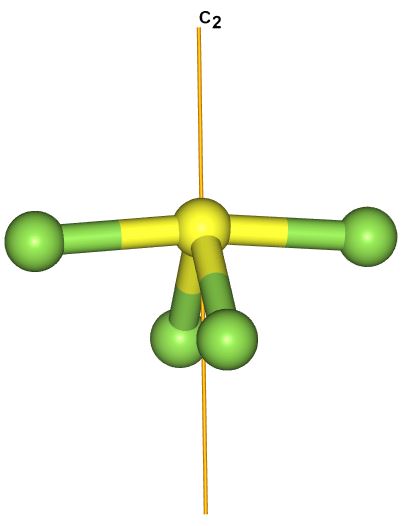
There are not 6 C5 axes.
There are not 3 C4 axes.
There are not 4 C3 axes.
The are no perpendicular C2 axes.
There is not an S4 axis.
There is not a perpendicular mirror plane.
There are however two mirror planes.
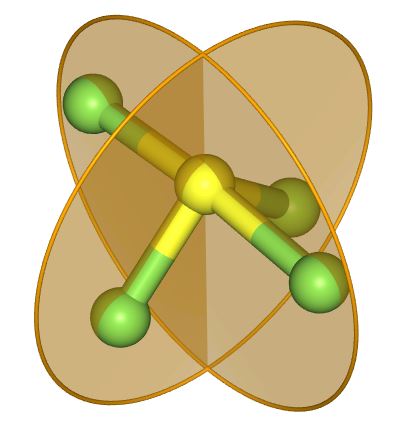
The point group of SF4 is C2v.
Once again we need the character table for the point group of SF4. Shown below are two different representations for the character table for the C2v point group. The first is the full character table. Although we ignored it in the methane example, this does include the d-orbital needed for hybridization. In the abbreviated character table the d-orbital has just been incorporated.
| C2v | E | C2 | σv (xz) | σv (yz) | Linear Functions | Quadratic Functions |
| A1 | +1 | +1 | +1 | +1 | z | x2, y2, z2 |
| A2 | +1 | +1 | -1 | -1 | Rz | xy |
| B1 | +1 | -1 | +1 | -1 | x, Ry | xz |
| B2 | +1 | -1 | -1 | +1 | z, Rx | yz |
Table \(\PageIndex{1}\): Full character table for the C2v point group.
| C2v | Orbitals |
| A1 | s, pz, dz2 |
| A2 | |
| B1 | px |
| B2 | py |
Table \(\PageIndex{2}\): Abbreviated character table for the C2v point group.
Exercise \(\PageIndex{4}\)
What are the symmetry labels for the orbitals on sulfur used to make SF4.
- Answer
-
s - a1
px - b1
py - b2
pz - a1
dz2 - a1
Step 3 - Making the LGOs
Before making the LGOs, we need to assign axis labels to the molecule. While there are many ways to do this, the proper convention is to assign the principle axis to be the z-axis (Figure \(\PageIndex{2}\)).
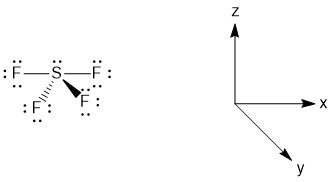
Figure \(\PageIndex{2}\). Assignment of the axes in SF4.
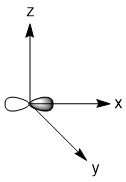
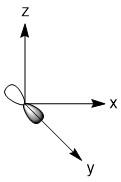
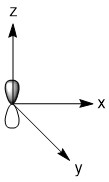
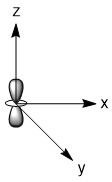
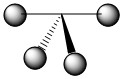
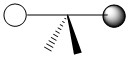
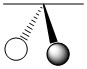
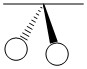
As mentioned in the previous section, we can treat terminal halides using this method. To make the pictures less cluttered, the halides will be represented just as the hydrogen atoms were and we will ignore the lone pair on the fluorine atoms. Based on the assigned axes above, the LGOs can be drawn to match the orbitals on the sulfur atom (Figure \(\PageIndex{3}\)).
 |
 |
 |
 |
 |
 |
 |
 |
 |
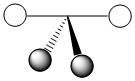 |
Figure \(\PageIndex{3}\). LGOs in SF4.
Step 3a - A few new twists
Before we go to step 4, we have a little bit of extra work to do. There are two features in this molecule that we have not dealt with before, the lone pair and the use of d-orbitals. Let's begin with the lone pair as this is something that could be encountered in molecules that do not break the octet rule. We know that this molecule has a lone pair on the sulfur atom. That lone pair has to be accounted for and must occupy one of the orbitals we considered as our basis functions. The question is, which orbital? The best option would be an orbital on the central atom that has no symmetry matched LGO. An example of this would be if all of the outer atoms in a molecule were sitting in the nodal plane of a p-orbital. We do not have that case in this example, so now we need to dig a little deeper. Our next best option is to consider all of the atomic orbitals of the central atom and ask which has the least amount of direct interaction with a LGO. The s-orbital on sulfur has interaction with all four fluorine atoms so it is not a good candidate. The p-orbitals all have interactions with two fluorine atoms, so they are slightly better options. In looking closer at the p-orbitals we see that the px orbital has very direct overlap with two fluorine atoms; the orbital is pointing directly at two atoms and both lobes interact. This is greater than the overlap of the py orbital in which the LGO does not lie directly on the y-axis. However, both lobes of the py orbital are interacting with the LGO. The pz orbital is somewhat similar in that the LGO does not lie along the z-axis. However, for this orbital, only one lobe of the sulfur pz is interacting with the LGO. So, given the options for the location of the lone pair, pz > py > px > s, we must assign the pz (a1) orbital to be the one with the lone pair. You may have noticed that the dz2 was not considered in the previous discussion. That is because the dz2 orbital is not accessible energetically. It is too high in energy to participate in any meaningful bonding in this molecule. While, we draw an LGO that would match this orbital, we do not consider any possible bonding or antibonding interaction between the LGO and the dz2 orbital. This is going to become very critical in the next step.
Step 4 - Constructing the MO diagram
In looking at the orbital energies from the previous section, we see that the 2s orbital of fluorine is MUCH lower in energy than the 2p of fluorine or the 3s and 3p of sulfur. It will not interact and does not need to be considered for this diagram. The 2p orbitals of fluorine fall between the 3s and 3p orbitals of sulfur. We will be bringing together a total of eight orbitals. Why only eight? Let's consider each of the combinations above (Figure \(\PageIndex{3}\)).
- Remember, we are ignoring the extra lone pairs on fluorine so that leaves a total of 12 orbitals that will not be shown in our diagram.
- The s-orbital (a1) from sulfur and the corresponding LGO will make a bonding and antibonding set.
- The px orbital (b1) orbital from sulfur and the corresponding LGO will make a bonding and antibonding set.
- The py orbital (b2) orbital from sulfur and the corresponding LGO will make a bonding and antibonding set.
- The pz orbital (a1) has been assigned as the orbital holding the lone pair so we ignore the LGO it could interact with. Note that this does not mean that the fluorine orbitals do not contribute to this orbital, but for the purpose of constructing the MO diagram we will ignore any contribution.
- The dz2 orbital (a1) of sulfur is not energetically accessible so we do not include it, but we do consider the LGO that would interact with it.
In sketching the MO diagram, the s-orbital of sulfur is the lowest energy orbital depicted and as such will give the lowest energy bonding MO and likely the highest energy antibonding MO. We will have two a1 orbitals that will be labeled as non-bonding (nb), one for the pz orbital with the lone pair, and one for the LGO that would match the dz2 orbital. As we are considering the orbitals to be localized on specific atoms, we will not change their energy from that of the atomic orbital from which they are derived. That leaves the b1 and b2 orbitals that will make a bonding and antibonding set. At first glance, you might expect these to be degenerate as they both are derived from p-orbitals on sulfur and the corresponding LGOs. However, upon examining the pictures above, (Figure \(\PageIndex{3}\)), it might be expected that the b1 bonding orbital might be slightly lower in energy than the b2 since there is a more direct overlap between the sulfur orbital and the LGO. For now we will treat the orbitals as being degenerate. This would lead to a MO diagram as pictured below (Figure \(\PageIndex{4}\)).
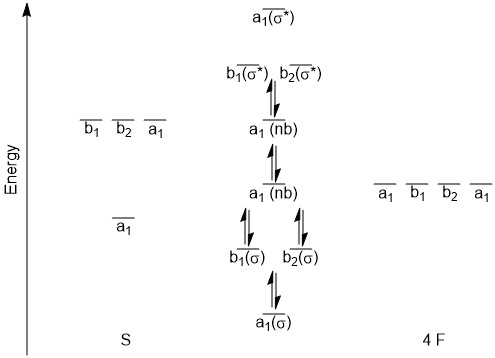
(Figure \(\PageIndex{4}\)). MO diagram for SF4.
Overall, this would give a bond order\( = \frac{6-0}{2}\ = 3\nonumber\) which is quite different from the Lewis structure. The bonding in structures of p-block elements with 'expanded' octets is not as straightforward as the Lewis structure might suggest. There are filled bonding orbitals that involve more than two atoms. There are also significant ionic contributions to what is typically drawn as covalent bonds.
How did we do?
Our MO diagram was generated using a pictorial method. Using computational methods it is possible to derive a more accurate MO diagram for this molecule. Show below are the orbitals and their energies as calculated by WebMO (Table \(\PageIndex{3}\)). Overall our MO diagram looks pretty good. We see a VERY slight difference in the energy of the b1 and b2 bonding orbitals. This difference is more pronounced in the antibonding orbitals. Based on the ABIMABTBIB principle, it is not surprising that we see a greater difference in the antibonding orbitals. So our approximation of the b1 and b2 orbitals as being degenerate is not unreasonable for the bonding MOs but not accurate for the antibonding.
You may also notice that all of the fluorine contributions are from p-orbitals. The 2s orbital of fluorine is much too low in energy to participate in these interactions. So the only orbital involved in bonding with the sulfur would be the p-orbital pointing directly at the sulfur atom. This justifies our treatment of the fluorine as being hydrogen-like. Again, we are ignoring any possible π-bonding interactions in this molecule.
We also estimated that the a1 (nb) would not change in energy much from the atomic orbitals they were derived from. The energy for the a1 (nb) derived from the sulfur pz is -12.049 eV which compares very favorably to the value of -12.0 eV for the 3p orbital of sulfur found in the previous page. For the a1 (nb) LGO, the energy was calculated to be -17.853 eV which is slightly higher than the -18.7 eV listed for the 2p orbitals of fluorine. Again, while not completely accurate, our pictorial method did a fairly reasonable job of generating the MO diagram.
(Table \(\PageIndex{3}\)). The MOs for SF4 (ignoring the lone pairs) as calculated and displayed by WebMO. The data for SF4 was imported into WebMO from the NIST Webbook as a computed 3-D structure and calculations were performed using B3LYP-6-31G(d).
| Orbital | Label | Energy (eV) |
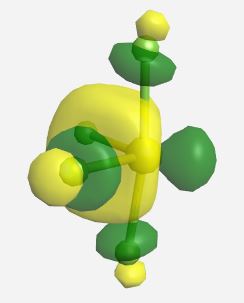 |
a1* | 0.517 |
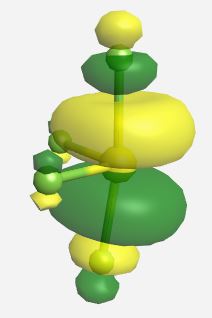 |
b1* | -1.950 |
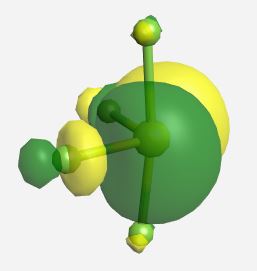 |
b2* | -4.083 |
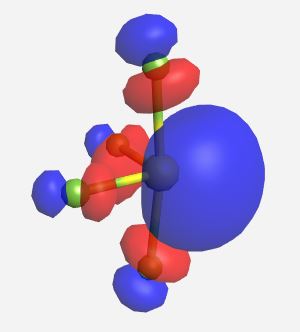 |
a1 (nb) | -12.049 |
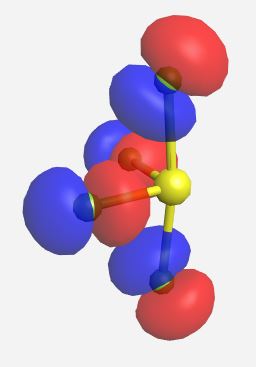 |
a1 (nb) | -17.853 |
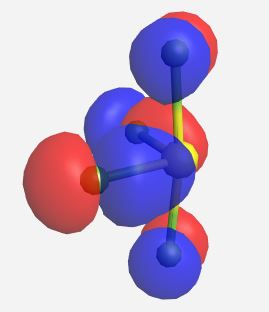 |
b2 | -18.559 |
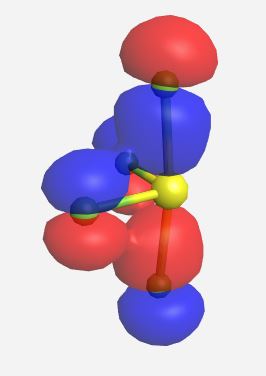 |
b1 | -18.697 |
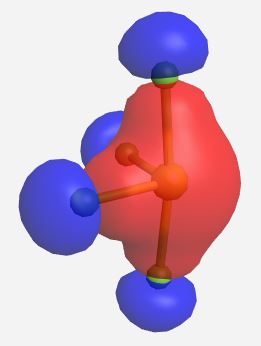 |
a1 | -20.655 |