
8.7: Bimolecular Elimination: E2
- Page ID
- 201163
E2, bimolecular elimination, was proposed in the 1920s by British chemist Christopher Kelk Ingold. Unlike E1 reactions, E2 reactions remove two subsituents with the addition of a strong base, resulting in an alkene.
Introduction
E2 reactions are typically seen with secondary and tertiary alkyl halides, but a hindered base is necessary with a primary halide. The mechanism by which it occurs is a single step concerted reaction with one transition state. The rate at which this mechanism occurs is second order kinetics, and depends on both the base and alkyl halide. A good leaving group is required because it is involved in the rate determining step. The leaving groups must be coplanar in order to form a pi bond; carbons go from sp3 to sp2 hybridization states.
To get a clearer picture of the interplay of these factors involved in a a reaction between a nucleophile/base and an alkyl halide, consider the reaction of a 2º-alkyl halide, isopropyl bromide, with two different nucleophiles. In one pathway, a methanethiolate nucleophile substitutes for bromine in an SN2 reaction. In the other (bottom) pathway, methoxide ion acts as a base (rather than as a nucleophile) in an elimination reaction. As we will soon see, the mechanism of this reaction is single-step, and is referred to as the E2 mechanism.
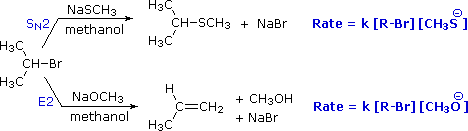
General Reaction
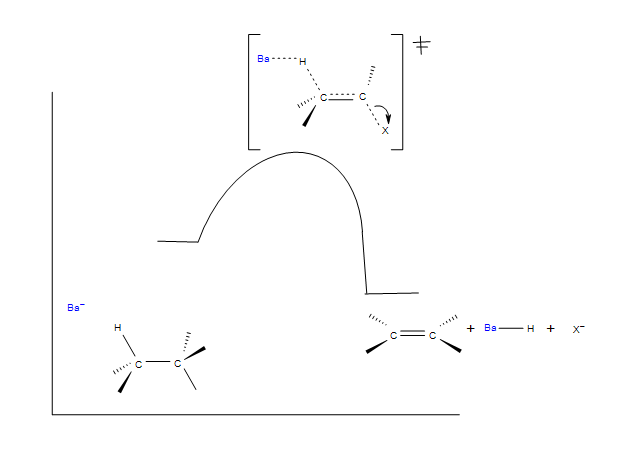
Below is a mechanistic diagram of an elimination reaction by the E2 pathway:
.

In this reaction Ba represents the base and Br representents a leaving group, typically a halogen. There is one transition state that shows the concerted reaction for the base attracting the hydrogen and the halogen taking the electrons from the bond. The product be both eclipse and staggered depending on the transition states. Eclipsed products have a synperiplanar transition states, while staggered products have antiperiplanar transition states. Staggered conformation is usually the major product because of its lower energy confirmation.
An E2 reaction has certain requirements to proceed:
- A strong base is necessary especially necessary for primary alkyl halides. Secondary and tertirary primary halides will procede with E2 in the precesence of a base (OH-, RO-, R2N-)
- Both leaving groups should be on the same plane, this allows the double bond to form in the reaction. In the reaction above you can see both leaving groups are in the plane of the carbons.
- Follows Zaitsev's rule, the most substituted alkene is usually the major product.
- Hoffman Rule, if a strically hindered base will result in the least substituted product.
E2 Reaction Coordinate
The Leaving Group Effect in E2 Reactions
As Size Increases, The Ability of the Leaving Group to Leave Increases: Here we revisit the effect size has on basicity. If we move down the periodic table, size increases. With an increase in size, basicity decreases, and the ability of the leaving group to leave increases. The relationship among the following halogens, unlike the previous example, is true to what we will see in upcoming reaction mechanisms.

Stereochemistry of the E2 Reaction
E2 elimination reactions of certain isomeric cycloalkyl halides show unusual rates and regioselectivity that are not explained by the principles thus far discussed. For example, trans-2-methyl-1-chlorocyclohexane reacts with alcoholic KOH at a much slower rate than does its cis-isomer. Furthermore, the product from elimination of the trans-isomer is 3-methylcyclohexene (not predicted by the Zaitsev rule), whereas the cis-isomer gives the predicted 1-methylcyclohexene as the chief product. These differences are described by the first two equations in the following diagram.
Unlike open chain structures, cyclic compounds generally restrict the spatial orientation of ring substituents to relatively few arrangements. Consequently, reactions conducted on such substrates often provide us with information about the preferred orientation of reactant species in the transition state. Stereoisomers are particularly suitable in this respect, so the results shown here contain important information about the E2 transition state.
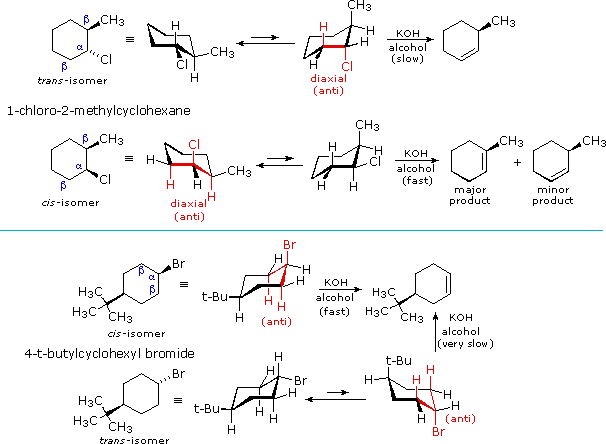
The most sensible interpretation of the elimination reactions of 2- and 4-substituted halocyclohexanes is that this reaction prefers an anti orientation of the halogen and the beta-hydrogen which is attacked by the base. These anti orientations are colored in red in the above equations. The compounds used here all have six-membered rings, so the anti orientation of groups requires that they assume a diaxial conformation. The observed differences in rate are the result of a steric preference for equatorial orientation of large substituents, which reduces the effective concentration of conformers having an axial halogen. In the case of the 1-bromo-4-tert-butylcyclohexane isomers, the tert-butyl group is so large that it will always assume an equatorial orientation, leaving the bromine to be axial in the cis-isomer and equatorial in the trans. Because of symmetry, the two axial beta-hydrogens in the cis-isomer react equally with base, resulting in rapid elimination to the same alkene (actually a racemic mixture). This reflects the fixed anti orientation of these hydrogens to the chlorine atom. To assume a conformation having an axial bromine the trans-isomer must tolerate serious crowding distortions. Such conformers are therefore present in extremely low concentration, and the rate of elimination is very slow. Indeed, substitution by hydroxide anion predominates.
 A similar analysis of the 1-chloro-2-methylcyclohexane isomers explains both the rate and regioselectivity differences. Both the chlorine and methyl groups may assume an equatorial orientation in a chair conformation of the trans-isomer, as shown in the top equation. The axial chlorine needed for the E2 elimination is present only in the less stable alternative chair conformer, but this structure has only one axial beta-hydrogen (colored red), and the resulting elimination gives 3-methylcyclohexene. In the cis-isomer the smaller chlorine atom assumes an axial position in the more stable chair conformation, and here there are two axial beta hydrogens. The more stable 1-methylcyclohexene is therefore the predominant product, and the overall rate of elimination is relatively fast.
A similar analysis of the 1-chloro-2-methylcyclohexane isomers explains both the rate and regioselectivity differences. Both the chlorine and methyl groups may assume an equatorial orientation in a chair conformation of the trans-isomer, as shown in the top equation. The axial chlorine needed for the E2 elimination is present only in the less stable alternative chair conformer, but this structure has only one axial beta-hydrogen (colored red), and the resulting elimination gives 3-methylcyclohexene. In the cis-isomer the smaller chlorine atom assumes an axial position in the more stable chair conformation, and here there are two axial beta hydrogens. The more stable 1-methylcyclohexene is therefore the predominant product, and the overall rate of elimination is relatively fast.
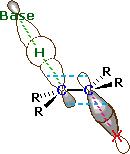
An orbital drawing of the anti-transition state is shown on the right. Note that the base attacks the alkyl halide from the side opposite the halogen, just as in the SN2 mechanism. In this drawing the α and β carbon atoms are undergoing a rehybridization from sp3 to sp2 and the developing π-bond is drawn as dashed light blue lines. The symbol R represents an alkyl group or hydrogen. Since both the base and the alkyl halide are present in this transition state, the reaction is bimolecular and should exhibit second order kinetics. We should note in passing that a syn-transition state would also provide good orbital overlap for elimination, and in some cases where an anti-orientation is prohibited by structural constraints syn-elimination has been observed.
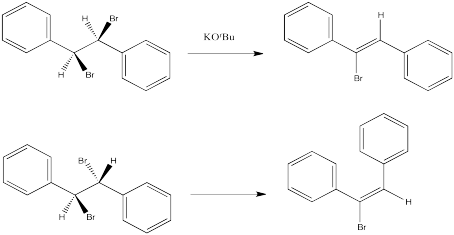
Instead, in an E2 reaction, stereochemistry of the double bond -- that is, whether the E or Z isomer results -- is dictated by the stereochemistry of the starting material, if it is diastereomeric. In other words, if the carbon with the hydrogen and the carbon with the halogen are both chiral, then one diastereomer will lead to one product, and the other diastereomer will lead to the other product.
The following reactions of potassium ethoxide with dibromostilbene (1,2-dibromo-1,2-diphenylethane) both occurred via an E2 mechanism. Two different diastereomers were used. Two different stereoisomers (E vs. Z) resulted.
Contributors
William Reusch, Professor Emeritus (Michigan State U.), Virtual Textbook of Organic Chemistry
Prof. Steven Farmer (Sonoma State University)