
9.2: Brønsted-Lowry Model
- Page ID
- 469952
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)The Brønsted-Lowry Acid Base Concept
The Brønsted-Lowry acid base concept overcomes the Arrhenius system's inability to describe reactions that take place outside of aqueous solution by moving the focus away from the solution and onto the acid and base themselves. It does this by redefining acid-base reactivity as involving the transfer of a hydrogen ion, \(H^+\), between an acid and a base. Specifically, a Brønsted acid is a substance that loses a \(H^+\) ion by donating it to a base. This means that a Brønsted base is defined as a substance which accepts \(H^+\) from an acid when it reacts.
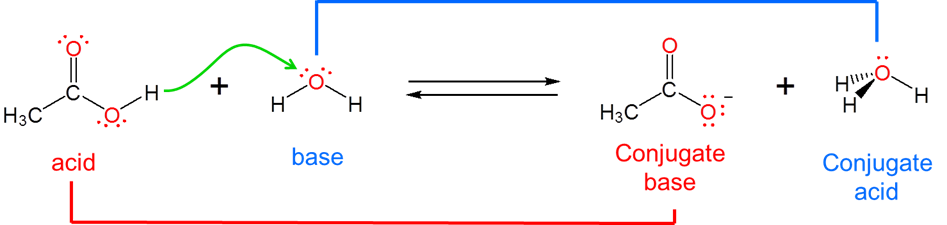
Because the Brønsted-Lowry concept is concerned with \(H^+\) ion transfer rather than the creation of a particular chemical species it is able to handle a diverse array of acid-base concepts. In fact, from the viewpoint of the Brønsted-Lowry concept, Arrhenius acids and bases are just a special case involving hydrogen ion donation and acceptance involving water. Arrhenius acids donate H+ ion to water, which acts as a Brønsted base to give H3O+
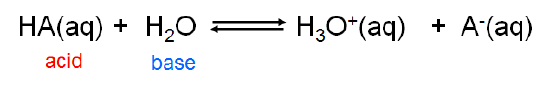 \[\label{ 6.3.1}\]
\[\label{ 6.3.1}\]
Similarly, Arrhenius bases act as Brønsted bases in accepting a hydrogen ion from the Brønsted acid water:
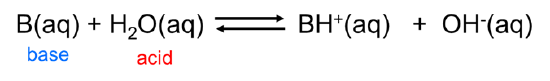 \[\]
\[\]
In this way it can be seen that Arrhenius acids and bases are defined in terms of their causing hydrogen ions to be donated to and abstracted from water, respectively, while Brønsted acids and bases are defined in terms of their ability to donate and accept hydrogen ions to and from anything.
Because the Brønsted-Lowry concept can handle any sort of hydrogen ion transfer it readily accommodates many reactions that Arrhenius theory cannot, including those that take place outside of water, such as the reaction between gaseous hydrochloric acid and ammonia:
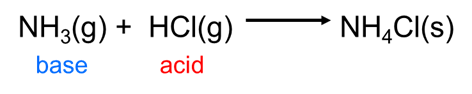 \[\]
\[\]
The classification of acids as strong or weak usually refers to their ability to donate or abstract hydrogen ions to or from water to give \(H_3O^+\) and \(OH^-\), respectively, - i.e. their Arrhenius acidity and basicity. However, acids and bases may be classified as strong and weak under the Brønsted-Lowry definition based on whether they completely transfer or accept hydrogen ions; it is just that in this case it is important to specify the conditions under which a given acid or base acts strong or weak. For example, acetic acid acts as a weak base in water but is a strong base in triethylamine, since in the latter case it completely transfers a hydrogen ion to triethylamine to give triethylammonium acetate. Alternatively, the acidity or basicity of a compound may be specified using a thermodynamic scale like the Hammett acidity.
Conjugate Acids and Bases
By redefining acids and bases in terms of hydrogen ion donation and acceptance the Brønsted-Lowry system makes it easy to recognize that when an acid loses its hydrogen ion it becomes a substance that is capable of receiving it back again, namely a base. Consider, for example, the base dissociation of ammonia in water. When ammonia acts as a Brønsted base and receives a hydrogen ion from water ammonium ion and hydroxide are formed:
 \[\]
\[\]
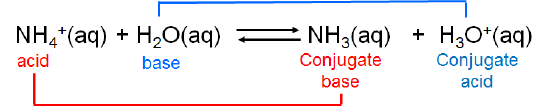
The ammonium ion is itself a weak acid that can undergo dissociation:
 \[\]
\[\]
In this case ammonia and ammonium ion are acid-base conjugates. In general acids and bases that differ by a single ionizable hydrogen ion are said to be conjugates of one another.
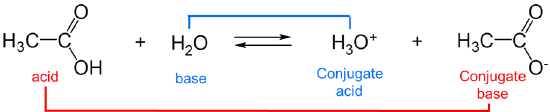
The strengths of conjugates vary reciprocally with one another so that the stronger the acid the weaker the base and vice versa. For example, in water, acetic acid acts as a weak Brønsted acid:
 \[\]
\[\]
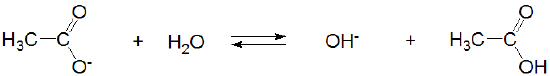
and acetic acid's conjugate base, acetate, acts as a weak Brønsted base.
 \[\]
\[\]
However, in liquid ammonia acetic acid acts as a strong Brønsted acid:
 \[\]
\[\]
while its conjugate base, acetate, is neutral.
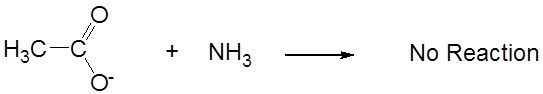 \[\]
\[\]
The reciprocal relationship between the strengths of acids and their conjugate bases has several consequences:
- Under conditions when an acid or base acts as a weak acid or base its conjugate acts as weak as well. Conversely, when an acid or base acts as a strong acid or base its conjugate acts as a neutral species.
- When a Brønsted acid and base react with one another the equilibrium favors formation of the weakest acid-base pair. That is why the acid-base reaction between acetic acid and ammonia in liquid ammonia proceeded to give the weak acid ammonium ion and neutral acetate. This consequence is particularly important for understanding the behavior of acids and bases in nonaqueous solvents, as illustrated by the following example.
Can a solution of lithium diisopropylamide in heptane be used to form lithium cyclopentadienide? The \(pK_a\) of cyclopentadiene and diisopropylamine are ~15 and 40, respectively, and the proposed reaction is as follows:
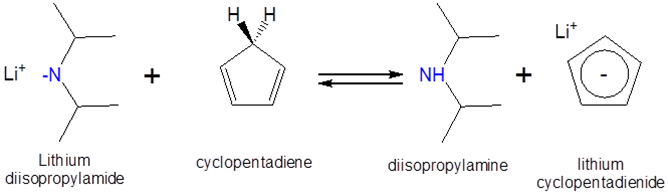
- Solution:
-
Since cyclopentadiene is a stronger acid than diisopropylamine (the stronger the acid the lower the \(pK_a\)) the equilibrium will favor protonation of the diisopropylamine by cyclopentadiene. Consequently addition of a heptane solution lithium diisopropylamide to monomeric cyclopentadiene should give lithium cyclopentadienide.
Molecular Structure and Brønsted Acidity and Basicity
Because the acidity of a given substance depends on the interplay between the relatively large values of its proton affinity and the energy associated with solvation of an acid's conjugate base it can be sometimes difficult to estimate the strength of an acid in a given solvent in the absence of detailed computations. Nevertheless a variety of simple ideas may be used to roughly estimate the relative strengths of acids. These should never be substituted for a detailed consideration of solvation but can serve as useful aids when thinking about trends and designing new Brønsted acids and bases.
Some simple factors that it can be helpful to consider when thinking about the strength of a given acid or base are:
Bond strength effects
The weaker the bond to the ionizable hydrogen, the stronger the acid. Strongly bonded hydrogen ions are difficult to remove, weakly bonded ones much less so.
Inductive effects
Inductive effects involve the donation or withdrawal of electrons from an atom by a group connected to it through bonds. Electron donating groups increase the electron density while electron withdrawing groups decrease it. Atoms or groups that withdraw electron density away from a center increase its acidity while those which donate electrons to the center decrease its acidity. The reasons for this follow from the heterolytic bond cleavage of acid ionization:
\[E-H→E:^-+ H^+\]
- When a bond to hydrogen is more polarized away from the H (more like\(^{-δ}E-H^{δ+}\)) it is easier to cleave off the hydrogen ion from that E-H bond. This may be seen from how the pKa values of acetic acid and its mono-, di-, and tri-chlorinated derivatives decreases with the extent of chlorination of the methyl group.

- Polarized E-H bonds also make for stronger Brønsted acids because the resulting \(E:^-\) conjugate base is more stable.
This leads to the third major factor that should be considered when thinking about acid strength.
Electronegativity effects
The more stable the acid's conjugate base, the stronger the Brønsted acid. All reactions are in theory reversible and so when considering the propensity of an acid to donate hydrogen ions it can be helpful to look at the reverse of hydrogen ion donation, namely protonation of the acid's conjugate base. If deprotonation of the acid gives a very stable conjugate base then deprotonation of the acid will be more favorable.
Two factors determine the stability of an acid's conjugate base.
- Conjugate bases in which a small amount of charge is on a large atom, spread over a large number atoms, and on electronegative atoms tend to be more stable. Conjugate bases in which a small amount of charge are spread over a large number of electronegative atoms are especially stable. That is why magic acid, a mixture of HF and \(SbF_5\), is so acidic; the single negative charge on its conjugate base is spread over six F atoms and on Sb in \(SbF_6^- \).
- Groups which tend to inductively polarize E-H bonds also tend to stabilize the conjugate base formed when that bond ionizes. In general, the more electronegative an atom, the better able it is to bear a negative charge. All other things being equal weaker bases have negative charges on more electronegative atoms; stronger bases have negative charges on less electronegative atoms. This is apparent from how inductive effects lead to an increase in the acidity of E-H bonds as the electronegativity of the element to which the acidic hydrogen is bound increases from left to right across a row of the periodic table. This horizontal periodic trend in acidity and basicity is apparent from the homologous series below in Figure \(\PageIndex{1}\).
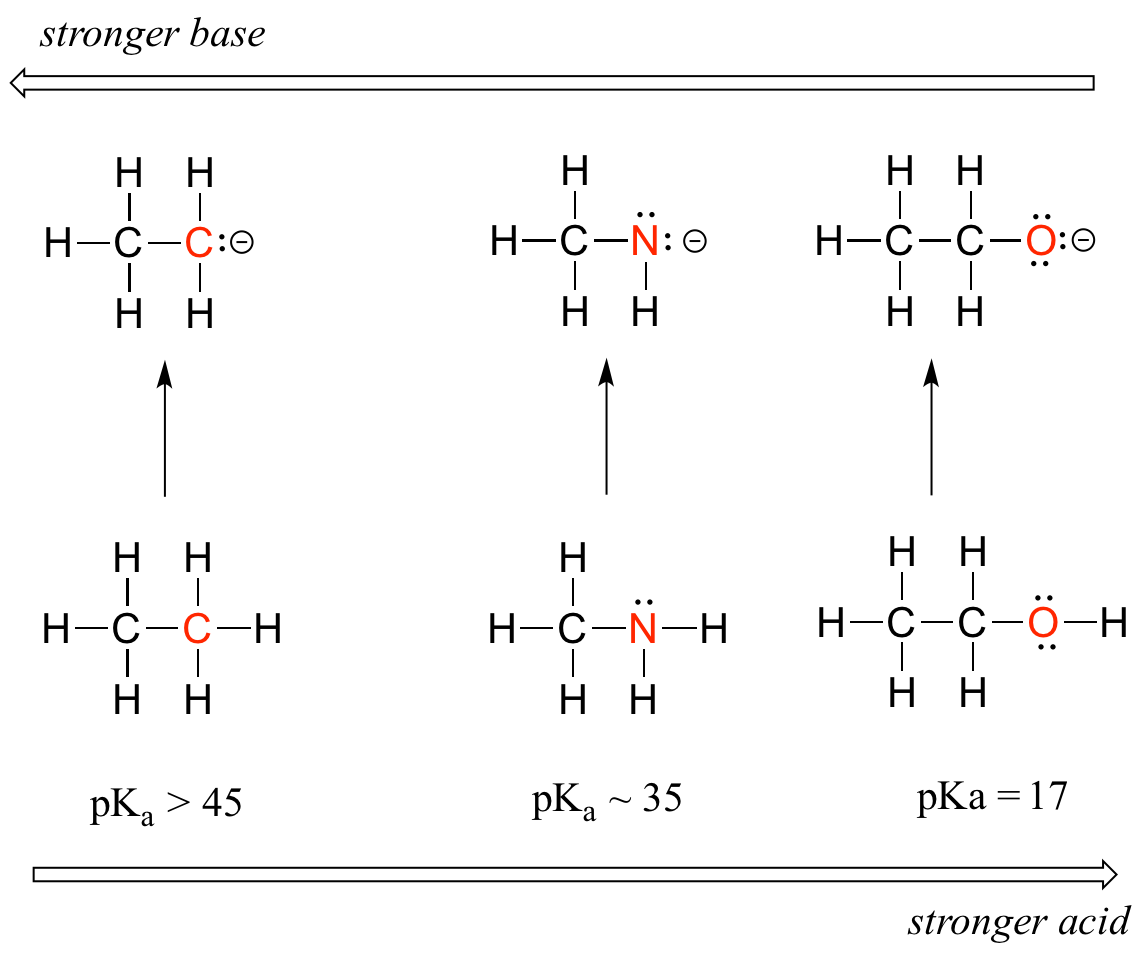
Notice how the inductive polarization of the E-H bond which results in greater acidity contributes to the greater stability of the conjugate base. For the case above look at where the negative charge ends up in each conjugate base. In the conjugate base of ethane, the negative charge is borne by a carbon atom, while on the conjugate base of methylamine and ethanol the negative charge is located on a nitrogen and an oxygen, respectively. Remember that electronegativity also increases as we move from left to right along a row of the periodic table, meaning that oxygen is the most electronegative of the three atoms, and carbon the least.
Thus, the methoxide anion is the most stable (lowest energy, least basic) of the three conjugate bases, and the ethyl carbanion anion is the least stable (highest energy, most basic). Conversely, ethanol is the strongest acid, and ethane the weakest acid.
Size effects
There are two classes of size effects to be considered:
- The larger the atom to which a H is bound in an E-H bond, the weaker the bond and the stronger the acid.
- Increased charge delocalization with increasing size. Electrostatic charges, whether positive or negative, are more stable when they are ‘spread out’ over a larger area. The greater the volume over which charge is spread in the acid's conjugate base, the more stable that base and the stronger the acid.
The impact of size effects are readily seen in the increase in acidity of the hydrogen halides, as illustrated by the vertical periodic trend in acidity and basicity below in Figure \(\PageIndex{2}\).
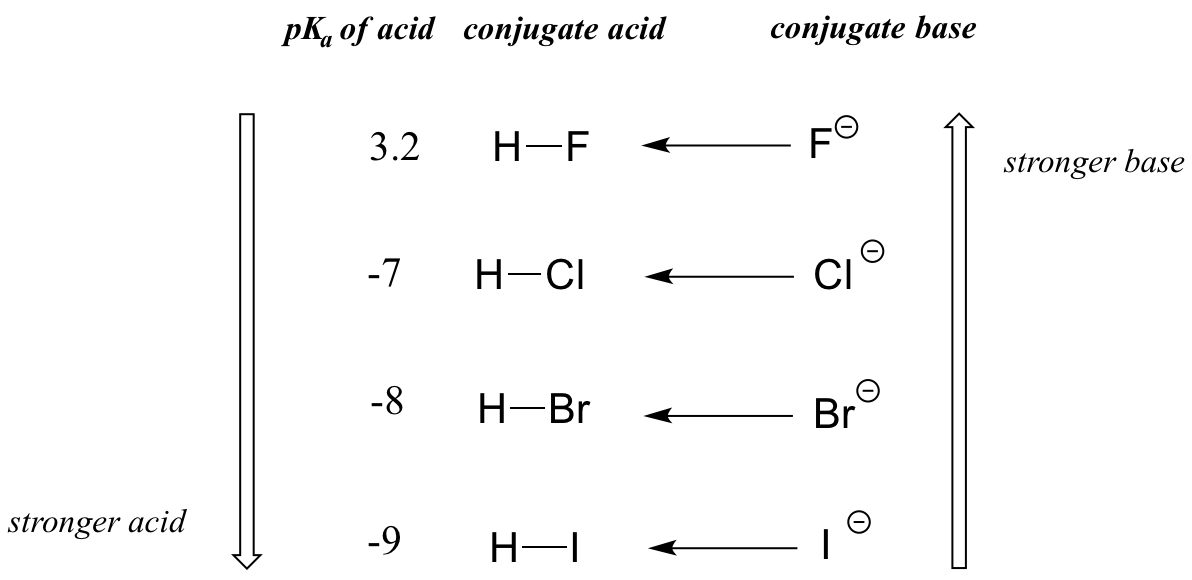
On going vertically down the halogen group from F to I the H-X bond strength decreases in the acid, making it easier to ionize, while the charge becomes more diffuse in the resultant X- ion, making the conjugate base more stable.
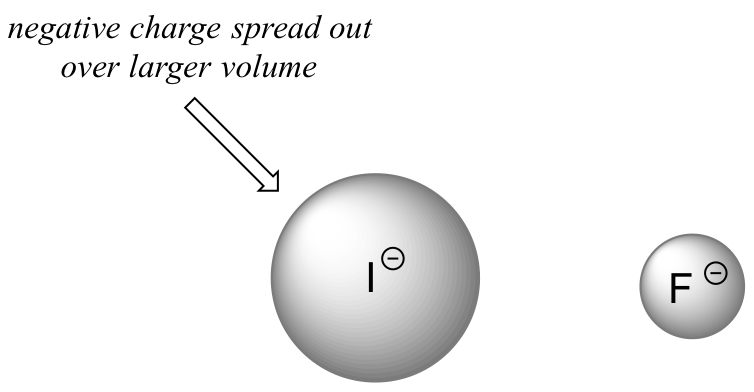
The increase in the acidity of the hydrogen halides down a group suggests that size effects are more important than inductive effects. In the case of the hydrogen halides because fluorine is the most electronegative halogen element, we might expect fluoride to also be the least basic halogen ion. But in fact, it is the least stable, and the most basic! It turns out that when moving vertically in the periodic table, the size of the atom trumps its electronegativity with regard to basicity. The atomic radius of iodine is approximately twice that of fluorine, so in an iodide ion, the negative charge is spread out over a significantly larger volume.
Now that we know how to quantify the strength of an acid or base, our next job is to gain an understanding of the fundamental reasons behind why one compound is more acidic or more basic than another. This is a big step: we are, for the first time, taking our knowledge of organic structure and applying it to a question of organic reactivity. Many of the ideas that we’ll see for the first here will continue to apply throughout the book as we tackle many other organic reaction types.
Periodic trends
First, we will focus on individual atoms, and think about trends associated with the position of an element on the periodic table. We’ll use as our first models the simple organic compounds ethane, methylamine, and ethanol, but the concepts apply equally to more complex biomolecules with the same functionalities, for example the side chains of the amino acids alanine (alkane), lysine (amine), and serine (alcohol).
Horizontal periodic trend in acidity and basicity:
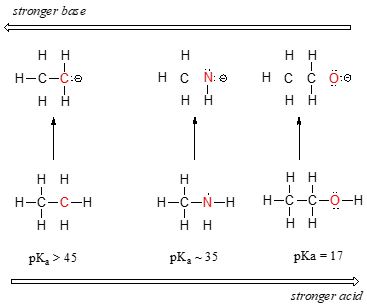
We can see a clear trend in acidity as we move from left to right along the second row of the periodic table from carbon to nitrogen to oxygen. The key to understanding this trend is to consider the hypothetical conjugate base in each case: the more stable (weaker) the conjugate base, the stronger the acid. Look at where the negative charge ends up in each conjugate base. In the conjugate base of ethane, the negative charge is borne by a carbon atom, while on the conjugate base of methylamine and ethanol the negative charge is located on a nitrogen and an oxygen, respectively. Remember from section 2.4A that electronegativity also increases as we move from left to right along a row of the periodic table, meaning that oxygen is the most electronegative of the three atoms, and carbon the least.
The more electronegative an atom, the better able it is to bear a negative charge. Weaker bases have negative charges on more electronegative atoms; stronger bases have negative charges on less electronegative atoms.
Thus, the methoxide anion is the most stable (lowest energy, least basic) of the three conjugate bases, and the ethyl carbanion anion is the least stable (highest energy, most basic). Conversely, ethanol is the strongest acid, and ethane the weakest acid.
When moving vertically within a given column of the periodic table, we again observe a clear periodic trend in acidity. This is best illustrated with the haloacids and halides: basicity, like electronegativity, increases as we move up the column.
Vertical periodic trend in acidity and basicity:
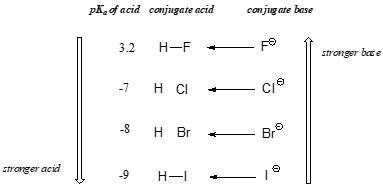
Conversely, acidity in the haloacids increases as we move down the column.
In order to make sense of this trend, we will once again consider the stability of the conjugate bases. Because fluorine is the most electronegative halogen element, we might expect fluoride to also be the least basic halogen ion. But in fact, it is the least stable, and the most basic! It turns out that when moving vertically in the periodic table, the size of the atom trumps its electronegativity with regard to basicity. The atomic radius of iodine is approximately twice that of fluorine, so in an iodide ion, the negative charge is spread out over a significantly larger volume:

This illustrates a fundamental concept in organic chemistry:
Electrostatic charges, whether positive or negative, are more stable when they are ‘spread out’ over a larger area.
We will see this idea expressed again and again throughout our study of organic reactivity, in many different contexts. For now, we are applying the concept only to the influence of atomic radius on base strength. Because fluoride is the least stable (most basic) of the halide conjugate bases, HF is the least acidic of the haloacids, only slightly stronger than a carboxylic acid. HI, with a \(pK_a\) of about -9, is almost as strong as sulfuric acid.
More importantly to the study of biological organic chemistry, this trend tells us that thiols are more acidic than alcohols. The \(pK_a\) of the thiol group on the cysteine side chain, for example, is approximately 8.3, while the \(pK_a\) for the alcohol group on the serine side chain is on the order of 17.
Remember the concept of 'driving force' that was introduced in section 6.2? Recall that the driving force for a reaction is usually based on two factors: relative charge stability, and relative total bond energy. Let's see how this applies to a simple acid-base reaction between hydrochloric acid and fluoride ion:
\[HCl + F^- \rightarrow HF + Cl^-\]
We know that HCl (\(pK_a\) -7) is a stronger acid than HF (\(pK_a\) 3.2), so the equilibrium for the reaction lies on the product side: the reaction is exergonic, and a 'driving force' pushes reactant to product.
What explains this driving force? Consider first the charge factor: as we just learned, chloride ion (on the product side) is more stable than fluoride ion (on the reactant side). This partially accounts for the driving force going from reactant to product in this reaction: we are going from less stable ion to a more stable ion.
What about total bond energy, the other factor in driving force? If you consult a table of bond energies, you will see that the H-F bond on the product side is more energetic (stronger) than the H-Cl bond on the reactant side: 570 kJ/mol vs 432 kJ/mol, respectively). This also contributes to the driving force: we are moving from a weaker (less stable) bond to a stronger (more stable) bond.
Resonance effects
In the previous section we focused our attention on periodic trends - the differences in acidity and basicity between groups where the exchangeable proton was bound to different elements. Now, it is time to think about how the structure of different organic groups contributes to their relative acidity or basicity, even when we are talking about the same element acting as the proton donor/acceptor. The first model pair we will consider is ethanol and acetic acid, but the conclusions we reach will be equally valid for all alcohol and carboxylic acid groups.
Despite the fact that they are both oxygen acids, the pKa values of ethanol and acetic acid are strikingly different. What makes a carboxylic acid so much more acidic than an alcohol? As before, we begin by considering the stability of the conjugate bases, remembering that a more stable (weaker) conjugate base corresponds to a stronger acid.
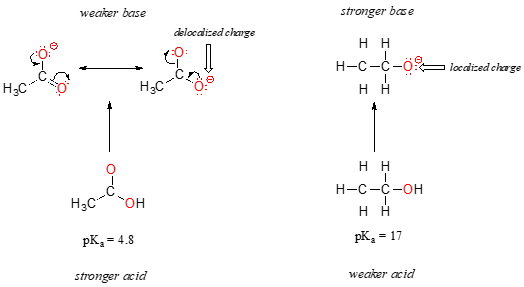
In both species, the negative charge on the conjugate base is located on oxygen, so periodic trends cannot be invoked. For acetic acid, however, there is a key difference: two resonance contributors can be drawn for the conjugate base, and the negative charge can be delocalized (shared) over two oxygen atoms. In the ethoxide ion, by contrast, the negative charge is localized, or ‘locked’ on the single oxygen – it has nowhere else to go. This makes the ethoxide ion much less stable.
Recall the important general statement that we made a little earlier: 'Electrostatic charges, whether positive or negative, are more stable when they are ‘spread out’ than when they are confined to one location.' Now, we are seeing this concept in another context, where a charge is being ‘spread out’ (in other words, delocalized) by resonance, rather than simply by the size of the atom involved.
The delocalization of charge by resonance has a very powerful effect on the reactivity of organic molecules, enough to account for the difference of over 12 \(pK_a\) units between ethanol and acetic acid (and remember, \(pK_a\) is a log expression, so we are talking about a factor of \(10^{12}\) between the \(K_a\) values for the two molecules!)
The resonance effect also nicely explains why a nitrogen atom is basic when it is in an amine, but not basic when it is part of an amide group. Recall that in an amide, there is significant double-bond character to the carbon-nitrogen bond, due to a minor but still important resonance contributor in which the nitrogen lone pair is part of a \(\pi \) bond.
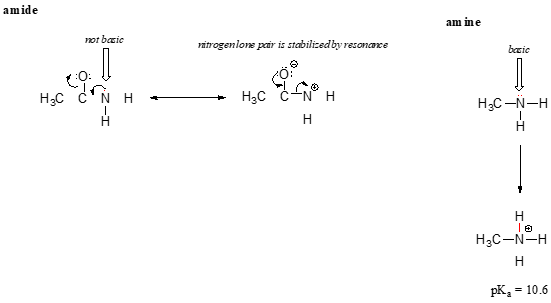
Whereas the lone pair of an amine nitrogen is ‘stuck’ in one place, the lone pair on an amide nitrogen is delocalized by resonance. Notice that in this case, we are extending our central statement to say that electron density – in the form of a lone pair – is stabilized by resonance delocalization, even though there is not a negative charge involved. Here’s another way to think about it: the lone pair on an amide nitrogen is not available for bonding with a proton – these two electrons are too ‘comfortable’ being part of the delocalized \(\pi \)-bonding system. The lone pair on an amine nitrogen, by contrast, is not so comfortable - it is not part of a delocalized \(\pi \) system, and is available to form a bond with any acidic proton that might be nearby.
If an amide group is protonated, it will be at the oxygen rather than the nitrogen.
- Draw the Lewis structure of nitric acid, \(HNO_3\).
- Nitric acid is a strong acid - it has a \(pK_a\) of -1.4. Make a structural argument to account for its strength. Your answer should involve the structure of the conjugate base of nitric acid.
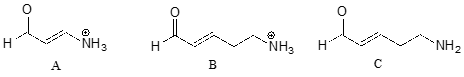
Rank the compounds below from most acidic to least acidic, and explain your reasoning.
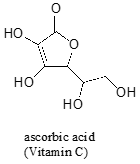
Often it requires some careful thought to predict the most acidic proton on a molecule. Ascorbic acid, also known as Vitamin C, has a \(pK_a\) of 4.1 - the fact that this is in the range of carboxylic acids suggest to us that the negative charge on the conjugate base can be delocalized by resonance to two oxygen atoms. Which if the four \(OH\) protons on the molecule is most acidic? Draw the structure of ascorbate, the conjugate base of ascorbic acid, then draw a second resonance contributor showing how the negative charge is delocalized to a second oxygen atom.
- Hint
-
Try deprotonating each \(OH\) group in turn, then use your resonance drawing skills to figure out whether or not delocalization of charge can occur.
Inductive effects
Compare the \(pK_a\) values of acetic acid and its mono-, di-, and tri-chlorinated derivatives:
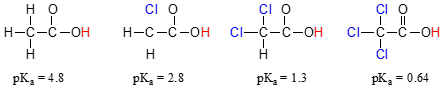
The presence of the chlorine atoms clearly increases the acidity of the carboxylic acid group, but the argument here does not have to do with resonance delocalization, because no additional resonance contributors can be drawn for the chlorinated molecules. Rather, the explanation for this phenomenon involves something called the inductive effect. A chlorine atom is more electronegative than a hydrogen, and thus is able to ‘induce’, or ‘pull’ electron density towards itself, away from the carboxylate group. In effect, the chlorine atoms are helping to further spread out the electron density of the conjugate base, which as we know has a stabilizing effect. In this context, the chlorine substituent can be referred to as an electron-withdrawing group. Notice that the \(pK_a\)-lowering effect of each chlorine atom, while significant, is not as dramatic as the delocalizing resonance effect illustrated by the difference in \(pK_a\) values between an alcohol and a carboxylic acid. In general, resonance effects are more powerful than inductive effects.
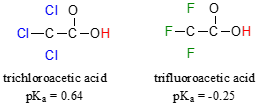
Because the inductive effect depends on electronegativity, fluorine substituents have a more pronounced \(pK_a\)-lowered effect than chlorine substituents.
In addition, the inductive takes place through covalent bonds, and its influence decreases markedly with distance – thus a chlorine two carbons away from a carboxylic acid group has a decreased effect compared to a chlorine just one carbon away.
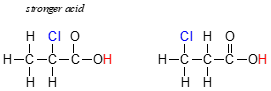
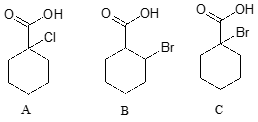
Rank the compounds below from most acidic to least acidic, and explain your reasoning.
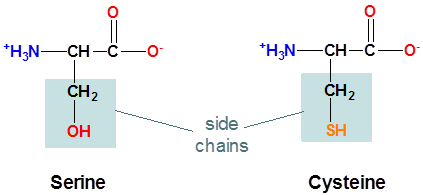
The structure of the amino acids serine and cysteine are shown below. Which do expect will have the more acidic side chain?
- Answer
-
Cysteine, since the cysteine side chain possesses an ionizable S-H bond while serine's side chain possesses an ionizable O-H bond. Since S is larger than O cysteine's S-H bond will be weaker than serine's O-H bond the and cysteine side chain's thiolate conjugate base more stable than the serine side chian's alkoxide conjugate base. In fact, the side chain \(pK_a\) of cysteine is 8.3 while serine is considered to be nonionizable under physiological conditions.
Binary Hydrides
The compounds formed between the elements and hydrogen are called binary hydrides. All such compounds can in principle act as Brønsted acids in reactions with a suitably strong base. However, as the electronegativity decreases down a group and increases from left to right across the periodic table the acidity of binary hydrides increases. In fact, on the left side of the periodic table the hydrides of extremely electropositive alkali and alkaline earth metals are not acidic but basic. They are perhaps best considered to be ionic salts of the hydride ion (\(\ce{H^{-}}\)). Consequently substances such as \(\ce{NaH}\) and \(\ce{CaH2}\) tend to act as Brønsted bases in their reactions.
\[\ce{NaH(s) + H2O(l) → Na^{+}(aq) + OH^{-}(aq) + H2(g)}\]
\[\ce{CaH2(s) + 2H2O(l) → Ca^{2+}(aq) + 2OH^{-}(aq) + 2H2(g)}\]
On the right side of the periodic table the binary hydrides of the nonmetals exhibit appreciable acidity.
\[\ce{HBr(aq) + H2O(l) → H^{+}(aq) + Br^{-}(aq)}\]
For this reason the binary nonmetal hydrides are termed acidic hydrides. Nevertheless not all are equally acidic. The dilute aqueous acid ionization constants for these hydrides are given in Figure \(\PageIndex{3}\). As can be seen from the constants in Figure \(\PageIndex{2}\) the ability of the hydrides to transfer a hydrogen to water increases across a period and down a group.
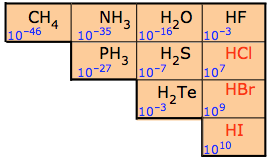
These trends are largely due to changes in the electronegativity and size of the nonmetal atom:
1. Going across a period the acid strength increases as there is an increase in electronegativity and the molecule gets more polar, with the hydrogen getting a larger partial positive charge. This makes it easier to heterolytically cleave the E-H bond to produce a stable anion.
\[E-H → E:^- + H^+\]
2. Going down a group the acid strength increases because the bond strength decreases as a function of increasing size of the nonmetal, and this has a larger effect than the electronegativity. In fact HF is a weak acid because it is so small that the hydrogen-fluorine bond is so strong that it is hard to break. Remember, the weaker the bond, the strong the acid strength. This is further illustrated in Table \(\PageIndex{1}\) where the weakest bond has produces the strongest acid.
| Relative Acid Strength | HF | << | HCl | < | HBr | < | HI |
|---|---|---|---|---|---|---|---|
| H–X Bond Energy (kJ/mol) | 570 | 432 | 366 | 298 | |||
| Ka | 10-3 | 107 | 109 | 1010 |
\[\
Oxyacids
Oxyacids (also known as oxoacids) are compounds of the general formula \(\ce{H_{n}EO_{m}}\), where \(\ce{E}\) is a nonmetal or early transition metal and the acidic hydrogens are attached directly to oxygen (not \(\ce{E}\)). This class of compounds includes such well-known acids as nitric acid, HNO3, and phosphoric acid, H3PO4.

The acidity of oxyacids follows three trends:
Electronegativity of the central atom
In a homologous series the acidity increases with the electronegativity of the central atom. Elements in the same group frequently form oxyacids of the same general formula. For example, chlorine, bromine, and iodine all form oxyacids of formula HOE: hypochlorous, hypobromous and hypoiodous acids. In the case of these homologous oxyacids the acidity is largely determined by the electronegativity of the central element. Central atoms which are better able to inductively pull electron density towards themselves make the oxygen-hydrogen bond that is to be ionized more polar and stabilize the conjugate base, OE-. Thus the acid strength in such homologous series increases with the electronegativity of the central atom. This may be seen from the data for the hypohalous acids in Table \(\PageIndex{2}\), in which the acid strength increases with the electronegativity of the halogen so that the order of acidity is:
HClO > HBrO > HIO
| HOE | Electronegativity of E | Ka |
|---|---|---|
| HOCl | 3.0 | 4.0 × 10−8 |
| HOBr | 2.8 | 2.8 × 10−9 |
| HOI | 2.5 | 3.2 × 10−11 |
Note this the influence of central atom electronegativity on the strength of oxyacids is exactly the opposite to that observed for the binary hydrides in Table \(PageIndex{5}\), for which acidity increased down a group giving the order of acidity:
\[\ce{HI>HBr>HCl \gg HF}\]
The reason for this is that in the hydrogen halides the bond to be broken (H-E bond) decreased in strength down the group while in oxyacids the bond to be broken is always an O-H bond and so varies much less in strength with the electronegativity of central atom.
Central element's oxidation state
For oxoacids of a given central atom the acidity increases with the central element's oxidation state or, in other words, the number of oxygens bound to the central atom. Here we are looking at the trend for acids in which there are variable numbers of oxygen bound to a given central atom. An examples is the perchloric (HClO4), chloric (HClO3), chlorous (HClO2), and hypochlorous (HClO) acid series. In such series the greater the number of oxygens the stronger the acid. This can be explained in several ways. From the viewpoint of the acid itself the key factor is again the inductive effect, in this case involving the ability of the oxygens attached to the central atom to pull on electron density across the OH bond. This is seen from the charge density diagram for the chlorine oxoacids shown in Figure \(\PageIndex{4}\), in which the partial positive charge on the acidic hydrogen increases with the number of oxygens present.
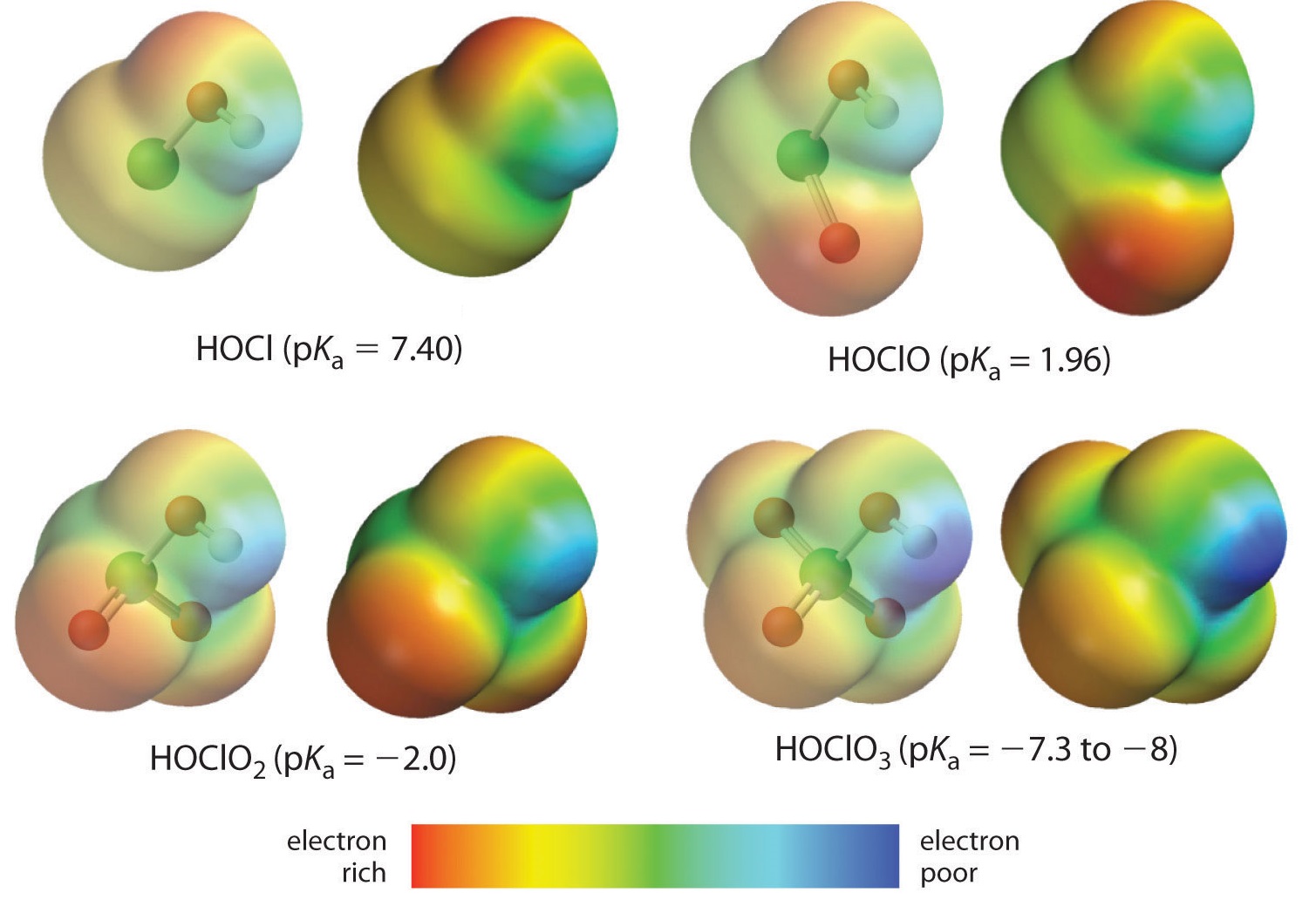
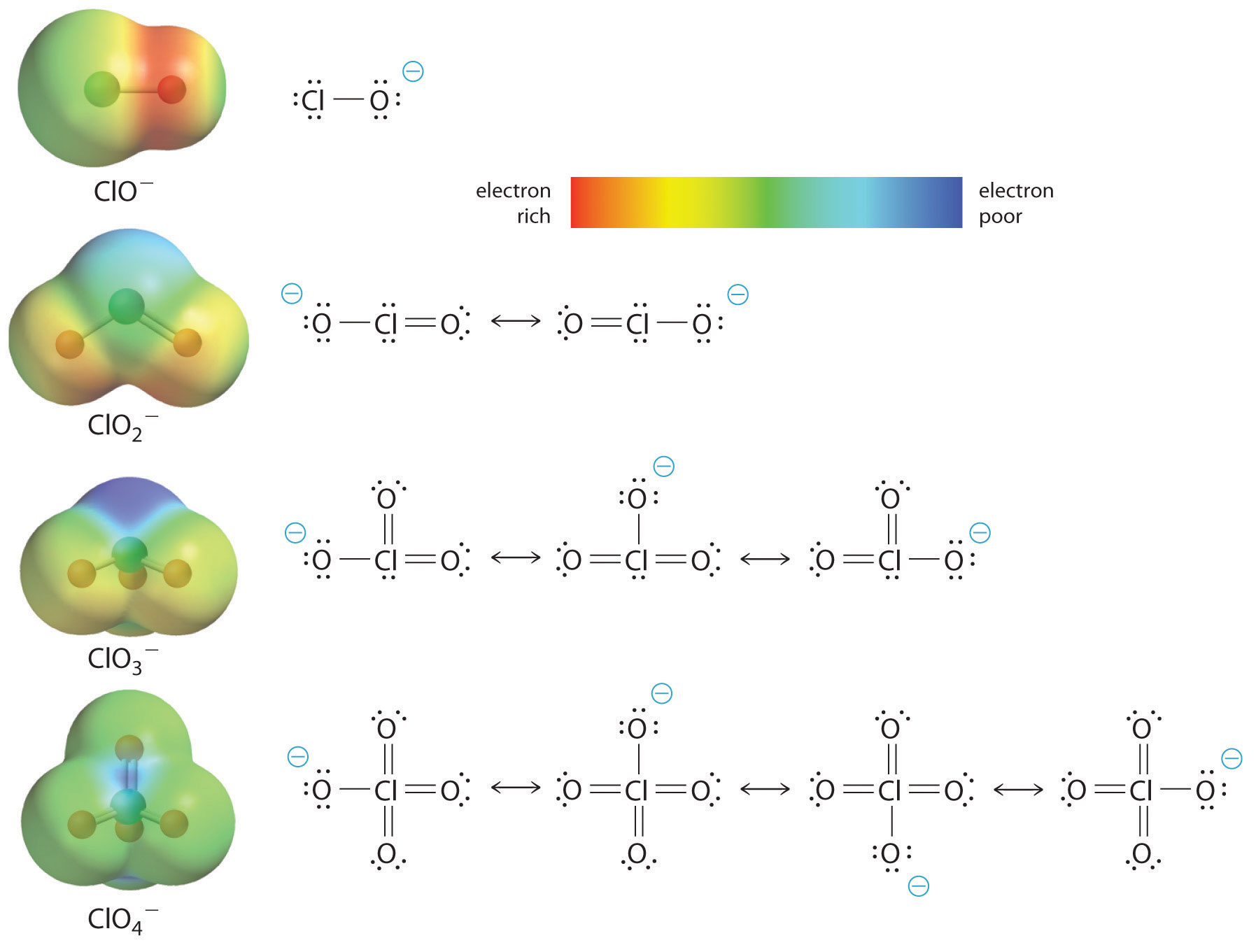
The increase in oxoacid acidity with the number of oxygens bound to the central atom may also be seen by considering the stability of the conjugate oxyanion. That the stability of the conjugate base increases with the number of oxygens may be seen from the charge distribution diagrams and Lewis bonding models for the chlorine oxyanions shown in figure \(\PageIndex{5}\). As the negative charge is spread over more oxygen atoms it becomes increasingly diffuse.
Sulfur and selenium both forms oxoacids of formula \(\ce{H_2EO_4}\) where E is either S or Se. These are called sulfurous and selenous acid, respectively. Which oxoacid would you expect to be more acidic: selenous acid or sulfurous acid?
- Answer
-
Sulfurous acid should be more acidic. Since sulfur is more electronegative than selenium sulfur will polarize OH bonds to a greater extent, making them more acidic. This prediction is borne out by a comparison of the \(pK_a\) values for the acids:
Acid \(pK_{a1}\) \(pK_{a2}\) sulfurous acid, \(H_2SO_3\) 1.85 7.2 selenous acid, \(H_2SeO_3\) 2.62 8.32
Successive proton removal
For polyprotic oxoacids the acidity decreases as each successive proton is removed. Oxoacids with multiple O-H bonds are called polyprotic since they can donate more than one hydrogen ion. In this case hydrogen ions are removed in successive ionization reactions. Examples include phosphoric and carbonic acid:
\[H_3PO_4 ⇌ H^+ + H_2PO_4^-~~~~~~~~~~~~~~~pK_{a1} = 2.2\]
\[H_2PO_4^- ⇌ H^+ + HPO_4^{2-}~~~~~~~~~~~~~~~pK_{a2} = 7.2\]
\[HPO_4 ⇌ H^+ + PO_4^{3-}~~~~~~~~~~~~~~~pK_{a3} = 12.4\]
\[H_2CO_3 ⇌ H^+ + HCO_3^-~~~~~~~~~~~~~~~pK_{a1} = 3.6\]
\[HCO_3^- ⇌ H^+ + CO_3^{2-}~~~~~~~~~~~~~~~pK_{a1} = 10.3\]
The dissociation constants for successive ionization constants decrease by about five orders of magnitude between successive hydrogen ions. This is reflected in Linus Pauling's rule for oxoacids and their oxyanions.
- The \(pK_a\) or an oxyacid of general formula \(\ce{E(OH)_{q}(O)_{p}}\) is given by \[pK_a = 8 - 5 \times p \label{PaulingRules}\]
- As an oxoaxid undergoes successive ionizations the \(pK_a\) increases by five each time.
The central theme of Pauling's Rules is that the more oxygen there are on the central atom, the more resonance structure that can be constructed of the conjugate base, which increases it stability and increases the acidity of the acid. However, as the acids successively ionize, they have fewer resonance structure. Pauling's Rules are phenomenological (i.e., not based on a theoretical basis). As with many empirical rules, they often work quite well, but they are approximations which may not work in all situations.
Calculate the theoretical \(pK_a\) values for phosphoric and carbonic acid and their dissociation produces and compare the results with the experimental \(pK_a\) values.
- Answer
-
For phosphoric acid, Pauling's rules (Equation \ref{PaulingRules}) predict the \(pK_a\) values quite well:
- \(H_3PO_4\): \(p = 3\) and \(q =1\) and \[pK_{a1, predicted} = 8 - 5 \times 1 = 3 \nonumber\] This slightly greater than the observed value of 2.2.
- \(H_2PO_4^-\): \[pK_{a2, predicted} = pK_{a1, experimental} + 5 = 7.2 \nonumber\] This Spot on with experiment.
- \(HPO_4^{2-}\): \[pK_{a3, predicted} = pK_{a2, experimental} + 5 = 12.2 \nonumber\]. This is slightly less than the experimental value of 12.4.
For carbonic acid Pauling's rules predict \(pK_{a1}\) reasonably well, but \(pK_{a2}\) less so:
- \(H_2CO_3\): \(p = 2\), \(q =1\) and \[pK_{a1, predicted} = 8 - 5 \times 1 = 3 \nonumber\] This is slightly lower than the observed value of 3.6.
- \(HCO_3^-\): \[pK_{a2, predicted} = pK_{a1, experimental} + 5 = 8.6 \nonumber\] This is 1.7 units less than the experimental value of 10.3.
In some cases discrepancies between experimental \(pK_a\) values and those predicted by Pauling's rules suggest that structural rearrangements may be taking place upon ionization or else that the reported \(pK_a\) values do not really represent the ionization in question because they do not fully account for all the equilibria taking place in solution. In the case of carbonic acid, however, the reason for the discrepancy between the predicted and experimental \(pK_{a2}\) values is not entirely clear.
Metal Ions as Acids
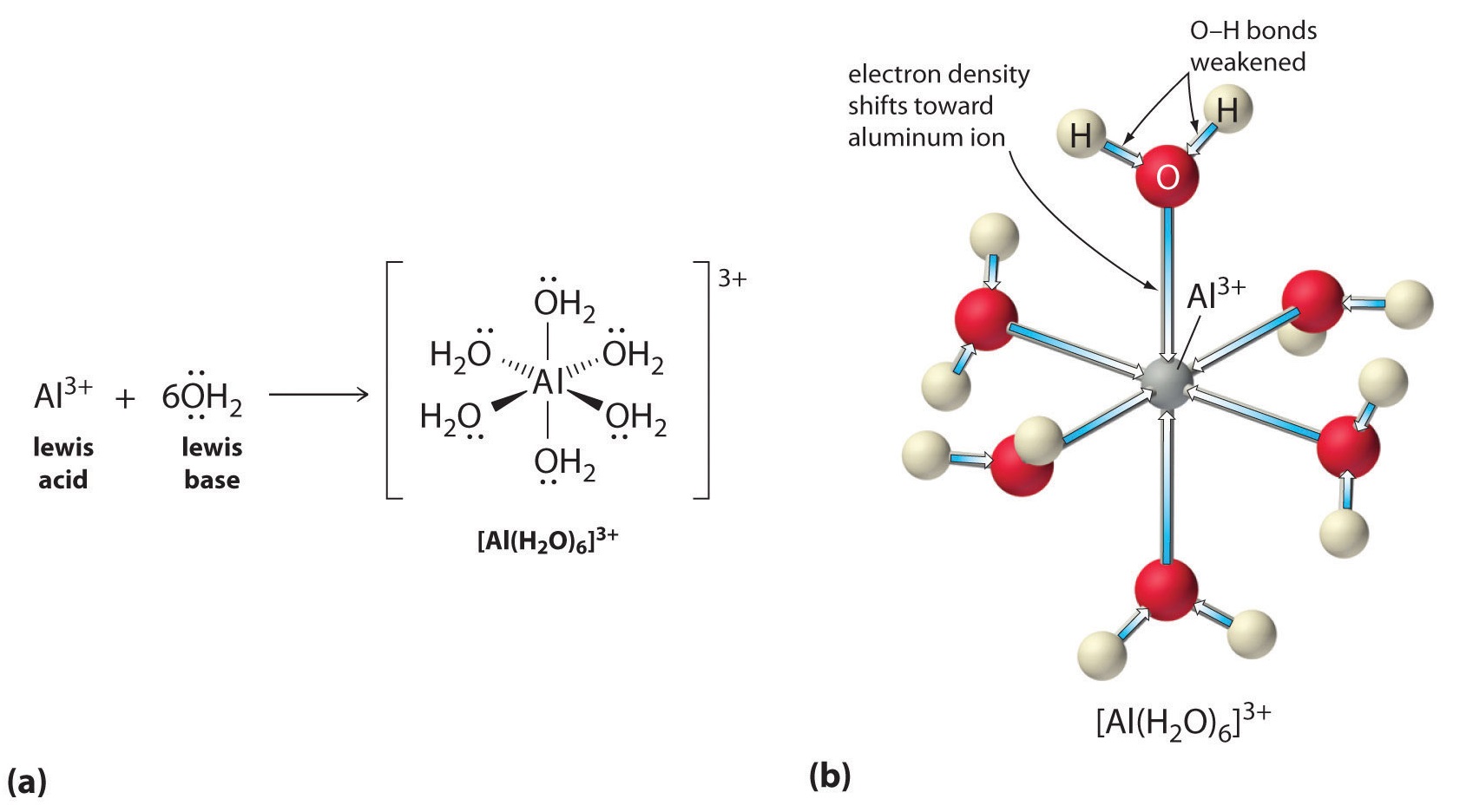
Aqueous solutions of simple salts of metal ions can also be acidic, even though a metal ion cannot donate a proton directly to water to produce \(H_3O^+\). Instead, a metal ion can act as a Lewis acid and interact with water, a Lewis base, by coordinating to a lone pair of electrons on the oxygen atom to form a hydrated metal ion.
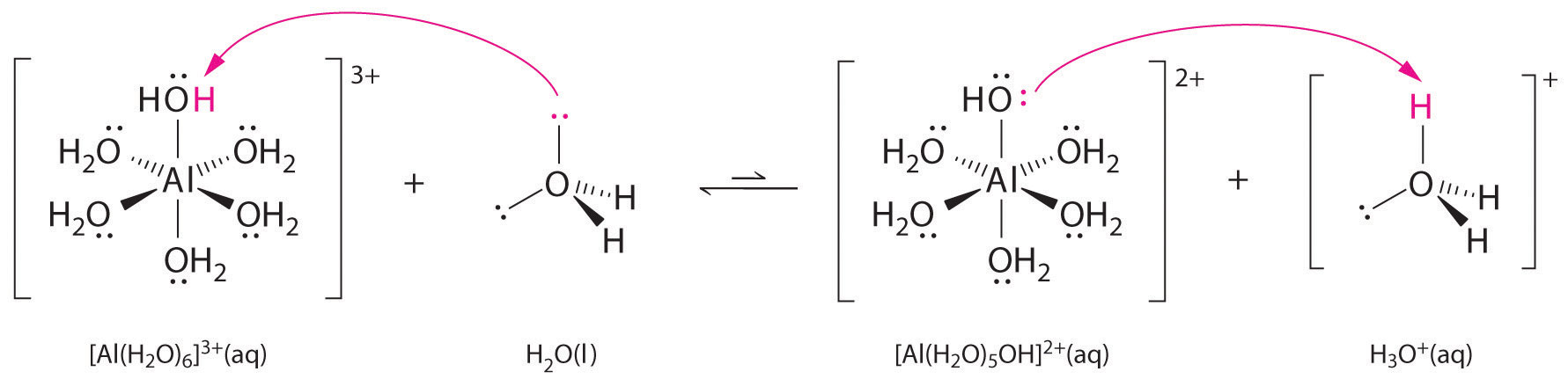
A water molecule coordinated to a metal ion is more acidic than a free water molecule for two reasons. First, repulsive electrostatic interactions between the positively charged metal ion and the partially positively charged hydrogen atoms of the coordinated water molecule make it easier for the coordinated water to lose a proton.
Second, the positive charge on the \(Al^{3+}\) ion attracts electron density from the oxygen atoms of the water molecules, which decreases the electron density in the \(\ce{O–H}\) bonds, as shown in Figure \(\PageIndex{6b}\). With less electron density between the \(O\) atoms and the H atoms, the \(\ce{O–H}\) bonds are weaker than in a free \(H_2O\) molecule, making it easier to lose a \(H^+\) ion. This is shown schematically in Figure \(\PageIndex{1}\).
Trends in Acidity
The acidity of a given metal ion largely depends on its charge to size ratio and electronegativity, although in some cases hardness and ligand field effects also play a role. The magnitude of this effect depends on the following factors, of which the first two are generally considered the most important.
The charge on the metal ion
A divalent ion (\(M^{2+}\)) has approximately twice as strong an effect on the electron density in a coordinated water molecule as a monovalent ion (\(M^+\)) of the same radius.
The radius of the metal ion
For metal ions with the same charge, the smaller the ion, the shorter the internuclear distance to the oxygen atom of the water molecule and the greater the effect of the metal on the electron density distribution in the water molecule.
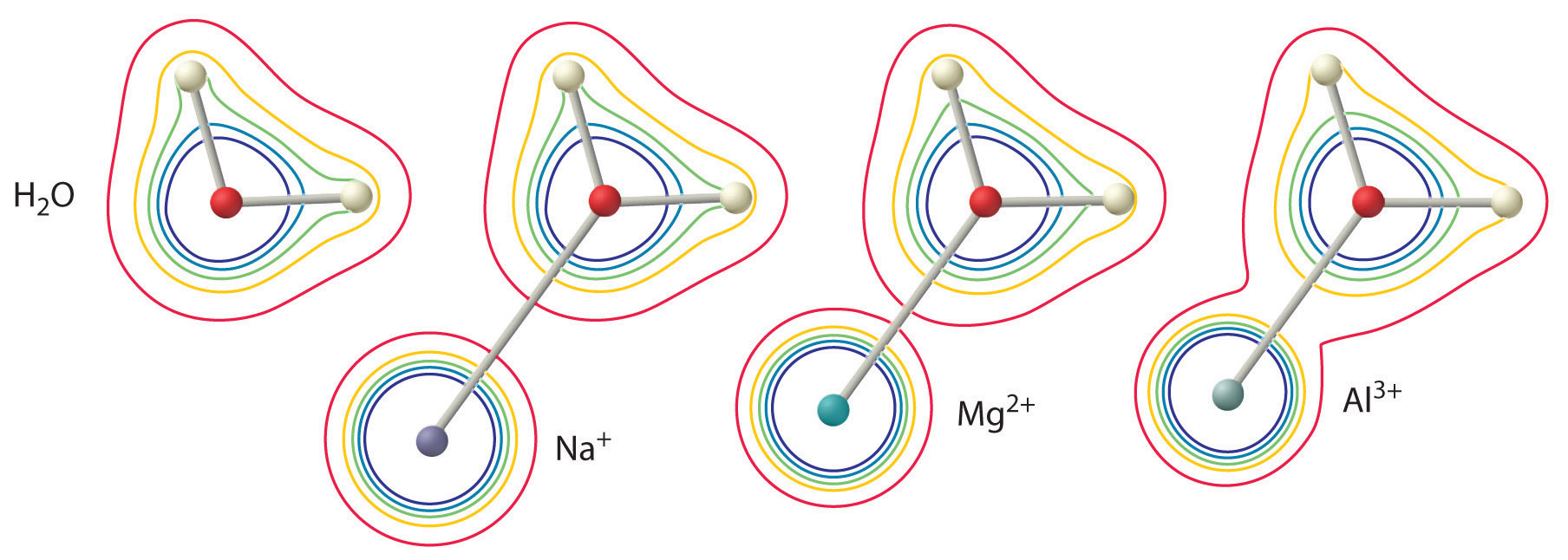
The first two of these factors explain why most alkali metal cations exhibit little acidity while aqueous solutions of small, highly charged metal ions, such as \(Al^{3+}\) and \(Fe^{3+}\), are acidic:
\[[Al(H_2O)_6]^{3+}_{(aq)} \rightleftharpoons [Al(H_2O)_5(OH)]^{2+}_{(aq)}+H^+_{(aq)} \label{16.36}\]
The \([Al(H_2O)_6]^{3+}\) ion has a \(pK_a\) of 5.0, making it almost as strong an acid as acetic acid. Because of the two factors described previously, the most important parameters for predicting the effect of a metal ion on the acidity of coordinated water molecules are the charge and ionic radius of the metal ion. Although the charge to size ratio is the simplest and most powerful predictor of metal ion acidity in water, three additional factors also can play a role:
Electronegativity
All other things being equal more electronegative elements are better able to withdraw electron density from a bound water ligand and consequently better at enhancing the ability of that water molecule to lose a hydrogen ion.
Hardness and Softness
Cation hardness or softness according to Pearson's Hard-Soft Acid Base Principle (HSAB). In general soft cations are more acidic than hard cations of the same charge and radius, as may be seen from the examples in Table \(\PageIndex{1}\). The greater than expected acidity of softer cations is thought to reflect the importance of covalent contributions to the metal-water bond.
| Cation | Classification | Radius (pm) | \(pK_a1\) |
|---|---|---|---|
| \(K^+\) | hard | 1.33 | 14 |
| \(Ag^+\) | soft | 1.26 | 10 |
| \(Mg^{2+}\) | hard | 0.65 | 12.2 |
| \(Cu^{2+}\) | soft | 0.69 | 7.3 |
| \(Ca^{2+}\) | hard | 0.99 | 12.6 |
| \(Cd^{2+}\) | soft | 0.97 | 9.0 |
| \(Sr^{2+}\) | hard | 1.13 | 13.1 |
| \(Hg^{2+}\) | soft | 1.10 | 3.6 |
Solvent Autoionization
The Brønsted-Lowry concept allows for an understanding of hydrogen ion transfer chemistry in amphoteric protic solvents. Amphoteric protic solvents are those which can both accept and receive hydrogen ions. From the viewpoint of the Brønsted-Lowry concept the acid-base chemistry in these solvents is governed by autoionization equilibria analogous to water autoionization.
\[2H_2O(l) ⇌ H_3O^+ + OH^-~~~~~~~~~~~~~~~~K_w = 1.0\times10^{-14}\]
For example, sulfuric acid ionizes according to the equation:
\[2H_2SO_4(l) ⇌ H_3SO_4^+ + HSO_4^-~~~~~~~~~~~~~~~K = 4\times10^{-4}\]
The magnitude of the solvent autoionization constant in a given amphoteric solvent determines the amount of protonated and deprotonated* solvent present. Since sulfuric acid's autoionization constant is much larger than that of water \(K_w = 10^{-14}\) the concentration of \(H_3SO_4^+\) and \(HSO_4^-\) present in pure sulfuric acid is ~0.02 M, much greater than the \(10^{-7}\) M \(H^+\) amd \(OH^-\) present in pure water.
In contrast, ammonia's autoionization constant is much less than that of water and only ~\(10^-14\) \{M NH_4^+\} and \{NH_2^-\} are present in pure ammonia.
\[2NH_3(l) ⇌ NH_4^+ + NH_2^-~~~~~~~~~~~~~~~~~K = 10^{-27}\]
Solvent Leveling Effect
The solvent leveling effect limits the strongest acid or base that can exist in acidic, basic, and amphoteric solvents. The conjugate acid and base of the solvent are the strongest Brønsted acid and bases that can exist in that solvent. To see why this is the case for acids consider the reaction between a Brønsted acid (HA) and solvent (S):
\[HA + S ⇌ HS^+ + A^-\]
This equilibrium will favor dissociation of whichever is a stronger acid - HA or HS+. If the acid is stronger it will mostly dissociate to give HS+, while if the solvent's conjugate acid is stronger the acid will be mostly un-ionized and remain as HA.
Any acid significantly stronger than HS+ will act as a strong acid and effectively dissociate completely to give the solvent's conjugate acid HS+. This also means that the relative acidity of acids stronger than HS+ cannot be distinguished in solvent S. This is called the leveling effect since the solvent "levels" the behavior of acids much stronger acids than it to that of complete dissociation. For example, there is no way to distinguish the acidity of strong acids like HClO4 and HCl in water since they both completely dissociate. However, it is possible to distinguish their relative acidities in solvents that are more weakly basic than the conjugate base of the strongest acid since then the acids will dissociate to different extents. Such solvents are called differentiating solvents. For example, acetonitrile (MeCN) acts as a differentiating solvent for HClO4 and HCl. Both HClO4 and HCl partially dissociate in MeCN, with the stronger acid HClO4 dissociating to a greater extent than HCl.
The leveling effect can also occur in basic solutions. The strongest Brønsted base, B, that can exist in a solvent is determined by the relative acidity of the solvent and the base's conjugate acid, BH+, determines whether the base will remain unprotonated and able to act as a base in that solvent. If the solvent is represented this time as HS then the relevant equilibrium is:
\[B + HS ⇌ BH^+ + S^-\]
The position of this equilibrium depends on whether B or S- is the stronger base. If the solvent's conjugate base, S- , is stronger then the base will remain unprotonated and available to act as a base. However, if B is a stronger base than S- it will deprotonate the solvent to give BH+ and S-. In this way the strongest base that can exist in a given solvent is the solvent's conjugate base. The relative strength of Brønsted bases can only be determined in solvents that are more weakly acidic than \(BH^+\); otherwise the bases will all be leveled to \(S^-\).
It is important to consider the levelling effect of protic solvents when performing syntheses that require the use of basic reagents. For instance, hydride and carbanion reagents (Lithium aluminum hydride, Grignard reagents, alklyllithium reagents, etc.) cannot be used as nucleophiles in protic solvents like water, alcohols, or enolizable aldehydes and ketones. Since carbanions are stronger bases than these solvent's conjugate bases they will instead act as Brønsted bases and deprotonate the solvent. For example if one adds n-butyllithium to water the result (along with much heat and possibly a fire) butane and a solution of lithium hydroxide will be obtained:
\[Li^+ + ^-:CH_2CH_2CH_2CH_3 + H_2O(l) → Li^+(aq) + OH^-(aq) + CH_3CH_2CH_2CH_3(g)\]
Since hydride and carbanion reagents cannot be used as nucleophiles in protic solvents like water or methanol they are commonly sold as solutions in solvents such as hexanes (for alklyllithium reagents) or tetrahydrofuran (for Grignard reagents and \(LiAlH_4\)).
References
- Wulfsberg, G. Principles of Descriptive Inorganic Chemistry University Science Books, 1991, pp. 28-30.
- (a) Gutmann, V. Allg. Prakt. Chem. 1970, 21, 116. (b) Gutmann, V. Fortschr. Chem. Forsch. 1972, 27, 59.
- Burgess, J. Metal Ions in Solution Ellis Horwood, 1978, pg. 268.
Contributors and Attributions
Stephen M. Contakes (Westmont College),
who expanded this section from a combination of
https://chem.libretexts.org/Bookshelves/General_Chemistry/Map%3A_General_Chemistry_(Petrucci_et_al.)/16%3A_Acids_and_Bases/16.7%3A_Ions_as_Acids_and_Bases