
16.10: Colligative Properties - Boiling-point Elevation
- Page ID
- 152704

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)The system we envision when we talk about boiling-point elevation is described schematically in Figure 7. We consider a solution of two components, \(A\) and \(B\). The mole fractions of \(A\) and \(B\), \(y_A\) and \(y_B\), specify the composition of the solution. We suppose that one of the components is present at a low concentration. We call this component the solute, and designate it as compound \(A\). Under these assumptions, we have \(y_A\approx 0\) and \(y_B=1-y_A\approx 1\). We assume further that \(A\) is nonvolatile, by which we mean that the vapor pressure of pure \(A\), \(P^{\textrm{⦁}}_A\), is very small. Then the second component, \(B\), comprises most of the material of the system. We call component \(B\) the solvent. We suppose that the \(A\)–\(B\) solution is in equilibrium with a gas phase. In principle, molecules of both components are present in this gas. Since we assume that essentially no component \(A\) is present in the gas phase, we have \(x_A=0\) and \(x_B=1\). We assume also that gas-phase \(B\) behaves as an ideal gas and solute \(A\) obeys Henry’s law.
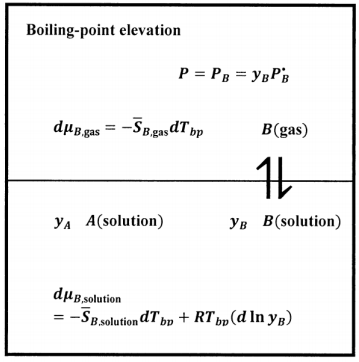
When we measure the boiling point of a liquid system, we find the temperature at which the vapor pressure of the system becomes equal to a specified value. For the normal boiling point, this pressure is 1 atmosphere, or 1.01325 bars. At the boiling point, liquid-phase solvent is in equilibrium with gas-phase solvent, so that the chemical potential of liquid-phase solvent is equal to the chemical potential of gas-phase solvent. That is, we have
\[\mu_{B,\mathrm{soluti}\mathrm{on}}=\mu_{B,\mathrm{gas}}.\nonumber \]
We want to describe the change in the equilibrium position that occurs when there is an incremental change in the solute concentration, \(dy_A\), while the pressure of the system remains constant. If the system is to remain at equilibrium, \(\mu_{B,\mathrm{solution}}=\mu_{B,\mathrm{gas}}\) must remain true. It follows that the chemical potentials of the two phases must change in tandem. Continued equilibrium implies that \(d\mu_{B,\mathrm{solution}}=d\mu_{B,\mathrm{gas}}\) when the solute concentration changes by \(dy_A\).
We can analyze the boiling-point elevation phenomenon for any fixed pressure at which pure liquid \(B\) can be at equilibrium with pure gas \(B\). Let us designate the fixed pressure as \(P^{\#}\). We designate the boiling-point temperature of pure solvent \(B\), at \(P^{\#}\), as \(T_B\); thus, \(P^{\#}=P^{\textrm{⦁}}_B\left(T_B\right)\), where \(P^{\textrm{⦁}}_B\left(T_B\right)\) designates the equilibrium vapor pressure of pure solvent \(B\) at temperature \(T_B\). Our goal is to find the temperature at which a binary solution is in equilibrium with pure gas \(B\) at the fixed pressure \(P^{\#}\). We let \(T_{bp}\) be the boiling temperature of the solution at \(P^{\#}\). The composition of the solution is specified by the solute concentration, \(y_A=1-y_B\).
Since we assume that the solute obeys Henry’s law, we choose the standard state for solute \(A\) to be the pure hypothetical liquid \(A\) whose vapor pressure is \({\textrm{ĸ}}_A\) at \(T\). We suppose that \({\textrm{ĸ}}_A\) is exceedingly small. From Section 16.4, we then have \(\tilde{a}_{A,\mathrm{solution}}=y_A\), so that
\[d ~ { \ln \tilde{a}_{A,\mathrm{solution}}\ }=d{ \ln y_A\ }\nonumber \]
at any temperature. From Section 16.8, we have
\[d ~ { \ln \tilde{a}_{B,\mathrm{solution}}\ }=d{ \ln y_B\ }\nonumber \]
Pure liquid-phase solvent is at equilibrium with gas-phase solvent at \(P^{\#}=P^{\textrm{⦁}}_B\left(T_B\right)\) and \(T_B\). We imagine that we create a solution by adding a small amount of solute \(A\), making the concentrations of solute and solvent \(y_A\) and \(y_B=1-y_A\), respectively. We maintain the system pressure constant at \(P^{\#}\), while changing the temperature to maintain equilibrium between gas-phase and solution-phase solvent \(B\). The new temperature is \(T_{bp}\).
The pressure of gas-phase B is constant at \(P^{\#}\). The temperature goes from \(T_B\) to \(T_{bp}\). We choose the activity standard state to be pure gas B at \(P^{\#}\) and T. This means that the activity of the pure gas is unity at every temperature, so that \(d{ \ln \tilde{a}_{B,\mathrm{gas}}\ }=0\).
It is worthwhile to note that we can arrive at this conclusion from a different perspective: From Section 14.14, the incremental change in the activity is
\[d ~ { \ln \tilde{a}_{B,\mathrm{gas}}\ }=\left(-\frac{\overline{H}_B}{RT^2}+\frac{\tilde{H}^o_B}{RT^2}\right)dT\nonumber \]
where \(\overline{H}_B\) is the partial molar enthalpy of gas-phase \(B\) at \(T\), and \(\tilde{H}^o_B\) is the partial molar enthalpy of \(B\) in its activity standard state at \(\ T\). Since we assume that the gas phase is essentially pure \(B\), we have \(\overline{H}_B=\tilde{H}^o_B\) and, again, \(d \ln \tilde{a}_{B,\mathrm{gas}}=0\).
From Section 14.3, we have the general result that
\[d\mu_B={\overline{V}}_BdP-{\overline{S}}_BdT+RT\left(d{ \ln \tilde{a}_B\ }\right)\nonumber \]
The system pressure and temperature are \(P=P^{\#}\) and \(T=T_{bp}\). For both the gas phase and the solution phase, we have \(dP=0\) and \(dT=dT_{bp}\). Since \(d{ \ln \tilde{a}_{B,\mathrm{gas}}\ }=0\), we have
\[d\mu_{B,\mathrm{gas}}=-{\overline{S}}_{B,\mathrm{gas}}dT_{bp}\nonumber \]
Since \(d ~ { \ln \tilde{a}_{B,\mathrm{solution}}\ }=d{ \ln y_B\ }\), we have
\[d\mu_{B,\mathrm{solution}}=-{\overline{S}}_{B,\mathrm{solution}}dT_{bp}+RT_{bp}\left(d{ \ln y_B\ }\right)\nonumber \]
The chemical potential of the pure, constant-pressure, gas-phase solvent depends only on temperature. The chemical potential of the constant-pressure, solution-phase solvent depends on temperature and solute concentration. Equilibrium is maintained if
\[d\mu_{B,\mathrm{solution}}=d\mu_{B,\mathrm{gas}}\nonumber \]
Substituting, we have
\[-{\overline{S}}_{B,\mathrm{solution}}dT_{bp}+RT_{bp}\left(d{ \ln y_B\ }\right)=-{\overline{S}}_{B,\mathrm{gas}}dT_{bp}\nonumber \]
Since \(y_B=1-y_A\approx 1\),
\[d{ \ln y_B\ }=d{ \ln \left(1-y_A\right)\ }={-dy_A}/{\left(1-y_A\right)}\approx -dy_A\nonumber \]
The relationship \(d\mu_{B,\mathrm{solution}}=d\mu_{B,\mathrm{gas}}\) becomes
\[-{\overline{S}}_{B,\mathrm{solution}}dT_{bp}-RT_{bp}dy_A=-{\overline{S}}_{B,\mathrm{gas}}dT_{bp}\nonumber \]
or
\[dy_A=\left(\frac{\overline{S}_{B,\mathrm{gas}}}-\overline{S}_{B,\mathrm{solution}} {RT_{bp}} \right)dT_{bp} \nonumber \]
We consider systems in which the boiling point of the solution, \(T_{bp}\), is little different from the boiling point of the pure solvent, \(T_B\). Then, \(T_B\approx T_{bp}\), and \({T_{bp}}/{T_B}\approx 1\). We let \(\Delta T=T_{bp}-T_B\), where \(\left|\Delta T\right|\ll T_B\). Since the solution is almost pure \(B\), the partial molar entropy of \(B\) in the solution is approximately that of pure \(B.\) Consequently, this partial molar entropy difference is, to a good approximation, just the entropy of vaporization of the solvent, at equilibrium, at the boiling point for the specified system pressure, \(P^{\#}=P^{\textrm{⦁}}_B\left(T_B\right)\). Then, since the vaporization of pure \(B\) at \(P^{\#}\) and \(T_B\) is a reversible process,
\[{\left({\overline{S}}_{B,\mathrm{gas}}-{\overline{S}}_{B,\mathrm{solution}}\right)}_{P^{\#},T_{bp}}\approx {\left({\overline{S}}^{\textrm{⦁}}_{B,\mathrm{gas}}-{\overline{S}}^{\textrm{⦁}}_{B,\mathrm{liquid}}\right)}_{P^{\#},T_B}=\Delta_{\mathrm{vap}}S_B={\Delta_{\mathrm{vap}}H_B}/{T_B}\nonumber \]
so that
\[dy_A=\left(\frac{\Delta_{\mathrm{vap}}H_B}{RT_{bp}T_B}\right)dT_{bp}\nonumber \]
In the solution, the solute mole fraction is \(y_A\); in the pure solvent, it is zero. At \(P^{\#}\) and \(T_B\), \(\Delta_{\mathrm{vap}}H_B\) is a constant. Integrating, between the limits \(\left(0,T_B\right)\) and \(\left(y_A,T_{bp}\right)\), we have
\[\int^{y_A}_0{dy_A}=\frac{\Delta_{\mathrm{vap}}H_B}{RT_B}\int^{T_{bp}}_{T_B}{\frac{dT_{bp}}{T_{bp}}}\nonumber \] and \[y_A=\frac{\Delta_{\mathrm{vap}}H_B}{RT_B}{ \ln \frac{T_{bp}}{T_B}\ }\nonumber \]
Introducing the approximation \({ \ln x\approx x-1\ }\), which is valid for \(x\approx 1\), we have
\[y_A=\frac{\Delta_{\mathrm{vap}}H_B}{RT_B}\left(\frac{T_{bp}}{T_B}-1\right)=\frac{\Delta_{\mathrm{vap}}H_B}{RT^2_B}\Delta T \nonumber \]
Solving for \(\Delta T\),
\[\Delta T=\left(\frac{RT^2_B}{\Delta_{\mathrm{vap}}H_B}\right)y_A\nonumber \]
Since the enthalpy of vaporization and the mole fraction are both greater than zero, \(\Delta T=T_{bp}-T_B>0\); that is, the addition of a non-volatile solute increases the boiling point of a liquid system. By measuring \(\Delta T\), we can find \(y_A\); if we know the molar mass of the solvent, we can calculate the number of moles of solute in the solution. If we know the mass of the solute used to prepare the solution, we can calculate the molar mass of the solute.
Frequently it is useful to express the solute concentration as a molality rather than a mole fraction. Using the dilute-solution relationship between mole fraction and molality from Section 16.6, \(y_A={\overline{M}_B{\underline{m}}_A}/{1000}\), the boiling-point elevation becomes:
\[\Delta T=\left(\frac{RT^2_B}{\Delta_{\mathrm{vap}}H_B}\right)\left(\frac{\overline{M}_B}{1000}\right){\underline{m}}_A\nonumber \]
Our theory predicts that the boiling-point elevation observed for a given solvent is proportional to the solute concentration and independent of the molecular characteristics of the solute. Experiments validate this prediction; however, its accuracy decreases as the solute concentration increases. Letting
\[{\textrm{ĸ}}_B=\frac{RT^2_B}{\Delta_{\mathrm{vap}}H_B}\nonumber \] and \[{\textrm{ĸ}}^*_B=\frac{RT^2_B\overline{M}_B}{1000\ \Delta_{\mathrm{vap}}H_B}\nonumber \]
we have \(\Delta T={\textrm{ĸ}}_By_A\) and \(\Delta T={\textrm{ĸ}}^*_B{\underline{m}}_A\). We call \({\textrm{ĸ}}_B\) or \({\textrm{ĸ}}^*_B\) the boiling-point (or boiling-temperature) elevation constant for solvent \(B\). For practical determination of molecular weights, we usually find \({\textrm{ĸ}}_B\) or \({\textrm{ĸ}}^*_B\) by measuring the increase in the boiling point of a solution of known composition.