
9.4: Chemical Bond Stability
- Page ID
- 13455
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)- Identify the nature of the energy of molecular orbitals of a diatomic as a function of intermolecular distance
- Identify the three integrals involved in calculation the total Molecular Orbital Energy: coulomb Integral, exchange integral, and overlap integral
As shown previously, we can construct two molecular orbitals for the \(\ce{H_2^{+}}\) system using the LCAO approximation with a basis set of two 1s atomic orbitals (i.e., the \(1s\) orbitals on hydrogen \(A\) (\(1s_A\)) and hydrogen \(B\) (\(1s_B\)):
\[| \psi _\pm\rangle = \dfrac{1}{\sqrt{2(1 \pm S )}} (1s_A \pm 1s_B) \label{9.3.7a} \]
The energy of these two molecular orbitals can be calculated from the expectation value integral of the Hamiltonian,
\[E_{\pm} = \left \langle \psi _{\pm} | \hat {H} _{elec} | \psi _{\pm} \right \rangle \label {9.4.1} \]
which can be expanded using the expanded molecular orbital wavefunctions in Equations \ref{9.3.7a} to give
\[E_{\pm} = \dfrac {1}{2(1 \pm S)} \left[ \underbrace{\left \langle 1s_A |\hat {H} _{elec} | 1s_A \right \rangle}_{H_{AA}} + \underbrace{\left \langle 1s_B |\hat {H} _{elec} | 1s_B \right \rangle }_{H_{BB}}\pm \underbrace{\left \langle 1s_A |\hat {H} _{elec} | 1s_B \right \rangle}_{H_{AB}} \pm \underbrace{\left \langle 1s_B |\hat {H} _{elec} | 1s_A \right \rangle}_{H_{BA}} \right] \label {9.4.2a} \]
where \(S\) is the overlap integral between the two atomic orbitals of the basis. The four integrals in Equation \(\ref{9.4.2a}\) can be represented by \(H_{AA}\), \(H_{BB}\), \(H_{AB}\), and \(H_{BA}\), respectively.
\[E_{\pm} = \dfrac {1}{2(1 \pm S)} \left[ H_{AA} + H_{BB} \pm H_{AB} \pm H_{BA} \right] \label {9.4.2b} \]
Show that Equation \(\ref{9.4.1}\) expands to give Equation \(\ref{9.4.2a}\) within the LCAO approximation that uses a basis set of only two 1s atomic orbitals.
- Answer
-
Here we have the wavefunction within the LCAO approximation that uses a basis set of only two 1s atomic orbitals (Equation \ref{9.3.7a}).
\[| \psi_{\pm} \rangle =\frac{1}{\sqrt{2(1 \pm S)}}\left(1_{S a} \pm 1_{S b}\right) \nonumber \]
And our LCAO approximation is equivalent to this if we plug in the wavefunction directly.
\[E_{\pm}= \langle \psi_{\pm}|\hat{H}| \psi_{\pm} \rangle =N^{2} \langle \left(1_{S a} \pm 1_{S b}\right)|\hat{H}|\left(1_{S a}-1_{S b}\right) \rangle \nonumber \]
We can see form above that the normalization constant squared results in:
\[N^{2}=\frac{1}{2(1 \pm S)} \nonumber \]
We now FOIL the mulitple (i.e., expaned term by term):
\[ \langle 1_{S a}|\hat{H}| 1_{S a} \rangle + \langle1_{S b}|\hat{H}| 1_{S b} \rangle \pm \langle 1_{S a}|\hat{H}| 1_{S b} \rangle \pm \langle 1_{S b}|\hat{H}| 1_{S a} \rangle \nonumber \]
Now we can see this is now equivalent to Equation \ref{9.4.2a} if the equation above is inserted (and adding a subscript to emphasize this only applies to the electronic wavefunction):
\[E_{\pm} = \dfrac {1}{2(1 \pm S)} \left[ \underbrace{\left \langle 1s_A |\hat {H} _{elec} | 1s_A \right \rangle}_{H_{AA}} + \underbrace{\left \langle 1s_B |\hat {H} _{elec} | 1s_B \right \rangle }_{H_{BB}}\pm \underbrace{\left \langle 1s_A |\hat {H} _{elec} | 1s_B \right \rangle}_{H_{AB}} \pm \underbrace{\left \langle 1s_B |\hat {H} _{elec} | 1s_A \right \rangle}_{H_{BA}} \right] \nonumber \]
Notice that \(A\) and \(B\) appear equivalently in the Hamiltonian operator for \(\ce{H_2^{+}}\). This equivalence means that integrals involving \(1s_A\) must be the same as corresponding integrals involving \(1s_B\), i.e.
\[ H_{AA} = H_{BB} \label {9.4.3} \]
and since the wavefunctions are real
\[ | A \rangle = \langle A | \nonumber \]
so
\[H_{AB} = H_{BA} \label {9.4.4} \]
These two equalities simplify Equation \ref{9.4.2b}:
\[ E_{\pm} = \dfrac {1}{1 \pm S} (H_{AA} \pm H_{AB}) \label {9.4.5} \]
Now examine the details of \(H_{AA}\) after inserting the Hamiltonian operator for \(\ce{H_2^{+}}\) (Equation 9.2.1):
\[H_{AA} = \underbrace{ \left \langle 1s_A \left| - \dfrac {\hbar ^2}{2m} \nabla ^2 - \dfrac {e^2}{4\pi \epsilon _0 r_A} \right| 1s_A \right \rangle }_{\color{red}{E_H}} + \dfrac {e^2}{4\pi \epsilon _0 R} \cancelto{ \color{red} {1}}{ \left \langle 1s_A | 1s_A \right \rangle} \underbrace{ - \left \langle 1s_A \left| \dfrac {e^2}{4 \pi \epsilon _0 r_B } \right| 1s_A \right \rangle}_{ \color{red} {J_{AB}}} \label {9.4.6} \]
- The first term is just the integral for the energy of the hydrogen atom of the 1s orbital, \(E_H\).
- The second integral is equal to 1 by normalization; the prefactor is just the Coulomb repulsion of the two protons.
- The last integral, including the minus sign, is represented by \(J\) and is called the Coulomb integral.
Physically \(J_{AB}\) is the potential energy of interaction of the electron located around proton \(A\) with proton \(B\). It is negative because it is an attractive interaction. It is the average interaction energy of an electron described by the \(1s_A\) function with proton \(B\).
The Coulomb Integral is the potential energy of electrostatic repulsion between the electron with the electron density in \(1S_A\) and the the electron with the electron density function \(1S_B\)
Now consider \(H_{AB}\).
\[H_{AB} = \underbrace{ \left \langle 1s_A \left| - \dfrac {\hbar ^2}{2m} \nabla ^2 - \dfrac {e^2}{4\pi \epsilon _0 r_B} \right| 1s_B \right \rangle}_{\color{red}{E_HS}} + \dfrac {e^2}{4\pi \epsilon _0 R} \cancelto{\color{red}{S}}{\left \langle 1s_A | 1s_B \right \rangle} - \underbrace{ \left \langle 1s_A \left| \dfrac {e^2}{4 \pi \epsilon _0 r_A } \right| 1s_B \right \rangle }_ {\color{red} {K_{AB}}}\label {9.4.7S} \]
- In the first integral we have the hydrogen atom Hamiltonian and the H atom function 1sB. The function 1sB is an eigenfunction of the operator with eigenvalue \(E_H\). Since \(E_H\) is a constant it factors out of the integral, which then becomes the overlap integral, \(S\). The first integral therefore reduces to \(E_HS\).
- The second term is just the Coulombic energy of the two protons times the overlap integral.
- The third term, including the minus sign, is given the symbol \(K\) and is called the exchange integral because the electron is described by the 1sA orbital on one side and by the 1sB orbital on the other side of the operator. The electron changes or exchanges position in the molecule.
In a coulomb integral, the electron always is in the same orbital; whereas, in an Exchange Integral, the electron is in one orbital on one side of the operator and in a different orbital on the other side.
Using the expressions for \(H_{AA}\) (Equation \ref{9.4.6}) and \(H_{AB}\) (Equation \ref{9.4.7S}) and substituting into Equation \(\ref{9.4.5}\) produces:
\[ \begin{align} E_{\pm} &= \dfrac {1}{1 \pm S} \left[ \left(E_H + \dfrac {e^2}{4\pi \epsilon_0 R}\right) (1 \pm S ) + J \pm K \right] \label {9.4.8} \\[4pt] &= E_H + \dfrac {e^2}{4\pi \epsilon _0 R} + \dfrac {J \pm K}{1 \pm S} \label {9.4.9} \end{align} \]
Equation \(\ref{9.4.9}\) tells us that the energy of the \(\ce{H_2^+}\) molecule is the energy of a hydrogen atom plus the repulsive energy of two protons plus some additional electrostatic interactions of the electron with the protons. These additional interactions are given by the last term
\[ \dfrac {J \pm K}{1 \pm S} \nonumber. \nonumber \]
If the protons are infinitely far apart then only \(E_H\) is nonzero, which we can set to zero by subtracting off:
\[ \begin{align} \Delta E_{\pm} &= E_{\pm} - E_H \\[4pt] &= \dfrac {e^2}{4\pi \epsilon _0 R} + \dfrac {J \pm K}{1 \pm S} \label {9.4.10} \end{align} \]
To get a chemical bond and a stable \(\ce{H_2^+}\) molecule \(\Delta E_{\pm}\) must be less than zero and have a minimum, i.e. \( \dfrac {J \pm K}{1 \pm S}\) must be sufficiently negative to overcome the positive repulsive energy of the two protons \(\dfrac {e^2}{4 \pi \epsilon _0R }\) for some value of \(R\). For large \(R\), these terms are zero, and for small \(R\), the Coulomb repulsion of the protons rises to infinity.
Show that Equation 9.2.1 follows from Equation \(\ref{9.4.5}\)
The Coulomb and Exchange Integrals
We will examine more closely how the Coulomb repulsion term and the integrals \(J\), \(K\), and \(S\) depend on the separation of the protons, but first we want to discuss the physical significance of \(J\), the Coulomb integral, and \(K\), the exchange integral. \(J\) and \(K\) have been defined as
\[ J_{AB} = \left \langle 1s_A \left| \dfrac {-e^2}{4 \pi \epsilon _0 r_B } \right|1s_A \right \rangle = - \int \varphi ^*_{1s_A} (r) \varphi _{1s_A} (r) \dfrac {e^2}{4 \pi \epsilon _0 r_B } d\tau \label {9.4.11} \]
\[ K_{AB} = \left \langle 1s_A \left| \dfrac {-e^2}{4 \pi \epsilon _0 r_A } \right|1s_B \right \rangle = - \int \varphi ^*_{1s_A} (r) \varphi _{1s_B} (r) \dfrac {e^2}{4 \pi \epsilon _0 r_A } d\tau \label {9.4.12} \]
Figure 9.4.2 shows graphs of the four terms contributing to the energy of \(\ce{H_2^{+}}\) (Equation \(\ref{9.4.10}\)). In Figure 9.4.2 , you can see that as the internuclear distance \(R\) approaches zero,
- the Coulomb repulsion of the two protons goes from near zero to a large positive number,
- the overlap integral goes for zero to one, and
- \(J\) and \(K\) become increasingly negative.
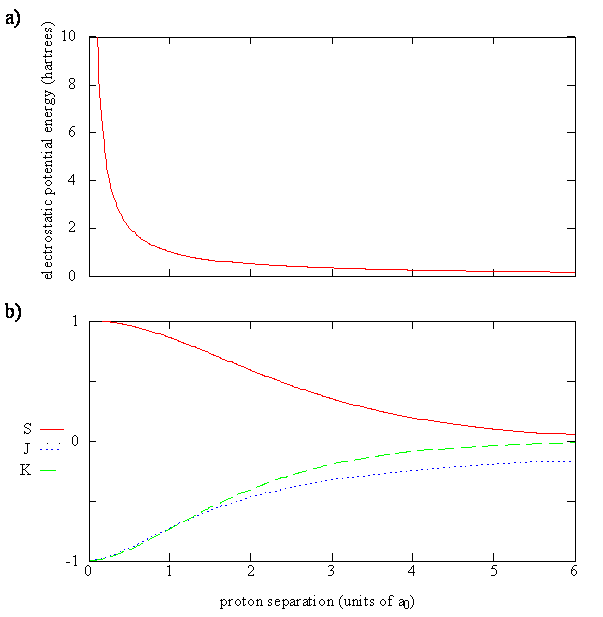
Note that both \(J\) and \(K\) integrals are negative since all quantities in the integrands of Equation \ref{9.4.11} and \ref{9.4.12} are positive. In the Coulomb integral, \(e \varphi ^*_{1s_A} (r) \varphi _{1a_A} (r)\) is the charge density of the electron around proton A, since r represents the coordinates of the electron relative to proton A. Since rB is the distance of this electron to proton B, the Coulomb integral gives the potential energy of the charge density around proton A interacting with proton B. \(J\) can be interpreted as an average potential energy of this interaction because \(e \varphi ^*_{1s_A} (r) \varphi _{1a_A} (r)\) is the probability density for the electron at point \(r\), and \(\dfrac {e^2}{4 \pi \epsilon _0 r_B }\) is the potential energy of the electron at that point due to the interaction with proton B. Essentially, J accounts for the attraction of proton B to the electron density of hydrogen atom A. As the two protons get further apart, this integral goes to zero because all values for rB become very large and all values for 1/rB become very small.
In the exchange integral, \(K\), the product of the two functions is nonzero only in the regions of space where the two functions overlap. If one function is zero or very small at some point then the product will be zero or small. The exchange integral also approaches zero as internuclear distances increase because the both the overlap and the \(1/r\) values become zero. The product \(e \varphi ^*_{1s_A} (r) \varphi _{1a_B} (r)\) is called the overlap charge density. Since the overlap charge density is significant in the region of space between the two nuclei, it makes an important contribution to the chemical bond. The exchange integral, \(K\), is the potential energy due to the interaction of the overlap charge density with one of the protons. While \(J\) accounts for the attraction of proton \(B\) to the electron density of hydrogen atom \(A\), \(K\) accounts for the added attraction of the proton due the build-up of electron charge density between the two protons.
Write a paragraph describing in your own words the physical significance of the Coulomb and exchange integrals for \(\ce{H_2^{+}}\).
Figure 9.4.3 shows the energy of H2+ relative to the energy of a separated hydrogen atom and a proton as given by Equation \(\ref{9.4.9}\). For the electron in the \(\psi_-\) orbital, the energy of the molecule, \(E_{el}(R)\), always is greater than the energy of the separated atom and proton.
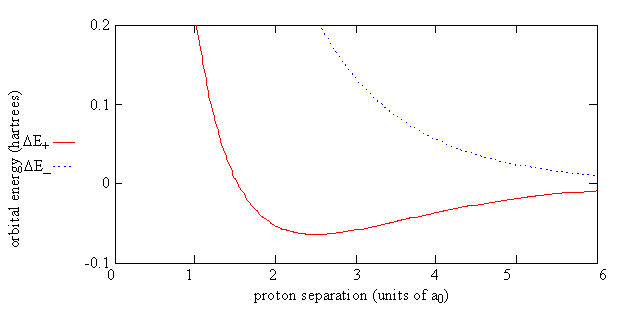
For the electron in the \(\psi _+\) orbital, you can see that the big effect for the energy of the bonding orbital, \(E_+(R)\), is the balance between the repulsion of the two protons \(\dfrac {e^2}{4 \pi \epsilon _0R }\) and J and K, which are both negative. J and K manage to compensate for the repulsion of the two protons until their separation is less than 100 pm (i.e the energy is negative up until this point), and a minimum in the energy is produced at 134 pm. This minimum represents the formation of a chemical bond. The effect of S is small. It only causes the denominator in Equation \(\ref{9.4.9}\) to increase from 1 to 2 as R approaches 0.
For the antibonding orbital, \(-K\) is a positive quantity and essentially cancels J so there is not sufficient compensation for the Coulomb repulsion of the protons. The effect of the -K in the expression, Equation \(\ref{9.4.9}\), for \(E_-\) is to account for the absence of overlap charge density and the enhanced repulsion because the charge density between the protons for \(\psi _-\) is even lower than that given by the atomic orbitals.
Contributors and Attributions
David M. Hanson, Erica Harvey, Robert Sweeney, Theresa Julia Zielinski ("Quantum States of Atoms and Molecules")