
8.2: Experimental Probes of Reaction Dynamics
- Page ID
- 11599

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Spectroscopic Methods
To follow the rate of any chemical reaction, one must have a means of monitoring the concentrations of reactant or product molecules as time evolves. In the majority of current experiments that relate to reaction dynamics, one uses some form of spectroscopic or alternative physical probe (e.g., an electrochemical signature or a mass spectrometric detection of product ions) to monitor these concentrations as functions of time. Of course, in all such measurements, one must know how the intensity of the signal detected relates to the concentration of the molecules that cause the signal. For example, in many absorption experiments, as illustrated in Figure 8.4, light is passed through a sample of thickness \(L\) and the intensity of the light beam in the absence of the sample \(I_0\) and with the sample present \(I\) are measured.
The Beer-Lambert law:
\[\log(I_0/I) = \varepsilon[A]L\]
then allows the concentration [A] of the absorbing molecules to be determined, given the path length \(L\) over which absorption occurs and given the extinction coefficient \(\varepsilon\) of the absorbing molecules.
These extinction coefficients, which relate to the electric dipole matrix elements as discussed in Chapter 6, are usually determined empirically by preparing a known concentration of the absorbing molecules and measuring the \(I_0/I\) ratio that this concentration produces in a cell of length \(L\). For molecules and ions that are extremely reactive, this calibration approach to determining \(\varepsilon\) is often not feasible because one cannot prepare a sample with a known concentration that remains constant in time long enough for the experiment to be carried out. In such cases, one often must resort to using the theoretical expressions given in Chapter 6 (and discussed in most textbooks on molecular spectroscopy) to compute \(\varepsilon\) in terms of the wave functions of the absorbing species. In any event, one must know how the strength of the signal relates to the concentrations of the species if one wishes to monitor chemical reaction or energy transfer rates.
Because modern experimental techniques are capable of detecting molecules in particular electronic and vibration-rotation states, it has become common to use such tools to examine chemical reaction dynamics on a state-to-state level and to follow energy transfer processes, which clearly require such state-specific data. In such experiments, one seeks to learn the rate at which reactants in a specific state \(\Phi_i\) react to produce products in some specific state \(\Phi_f\). One of the most common ways to monitor such state-specific rates is through a so-called pump-probe experiment in which
i. A short-duration light pulse is used to excite reactant molecules to some specified initial state \(\Phi_i\). Usually a tunable laser is used because its narrow frequency spread allows specific states to be pumped. The time at which this pump laser thus prepares the excited reactant molecules in state \(\Phi_i\) defines \(t = 0\).
ii. After a delay time of duration t, a second light source is used to probe the product molecules that have been formed in various final states, \(\Phi_f\). Often, the frequency of this probe source is scanned so that one can examine populations of many such final states.
The concentrations of reactant and products molecules in the initial and final states \(\Phi_i\) and \(\Phi_f\) are determined by the Beer-Lambert relation assuming that the extinction coefficients \(\varepsilon_i\) and \(\varepsilon_f\) for these species and states absorption are known. In the former case, the extinction coefficient \(\varepsilon_i\) relates to absorption of the pump photons to prepare reactant molecules in the specified initial state. In the latter, \(\varepsilon_f\) refers to absorption of the product molecules that are created in the state \(\Phi_f\). Carrying out a series of such final-state absorption measurements at various delay times t allows one to determine the concentration of these states as a function of time.
This kind of laser pump-probe experiment is used not only to probe specific electronic or vibration/rotation states of the reactants and products but also when the reaction is fast (i.e., complete in 10‑4s or less). In these cases, one is not using the high frequency resolution of the laser but its fast time response. Because laser pulses of quite short duration can be generated, these tools are well suited in such fast chemical reaction studies. The reactions can be in the gas phase (e.g., fast radical reactions in the atmosphere or in explosions) or in solution (e.g., photo-induced electron transfer reactions in biological systems).
Beam Methods
Another approach to probing chemical reaction dynamics is to use a beam of reactant molecules A that collides with other reactants B that may also in a beam or in a bulb in equilibrium at some temperature T. Such crossed-beam and beam-bulb experiments are illustrated in Figure 8.5.

Almost always, these beam and bulb samples contain molecules, radicals, or ions in the gas phase, so these techniques are most prevalent in gas-phase dynamics studies.
The advantages of the crossed-beam type experiments are that:
- one can control the velocities, and hence the collision energies, of both reagents,
- one can examine the product yield as a function of the angle \(\theta\) through which the products are scattered,
- one can probe the velocity of the products and,
- by using spectroscopic methods, one can determine the fraction of products generated in various internal (electronic/vibrational/rotational) states.
Such measurements allow one to gain very detailed information about how the reaction rate coefficient depends on collisional (kinetic) energy and where the total energy available to the products is deposited (i.e., into product translational energy or product internal energy). The angular distribution of product molecules can also give information about the nature of the reaction process. For example, if the A + B collision forms a long-lived (i.e., on rotational time scales) collision complex, the product C molecules display a very isotropic angular distribution. In contrast, reactions that proceed more impulsively show product angular distributions that are either strongly back-scattered or strongly forward-scattered rather than isotropic.
In beam-bulb experiments, one is not able to gain as much detailed information because one of the reactant molecules B is not constrained to be moving with a known fixed velocity in a specified direction when the \(A + B \rightarrow C\) collisions occur. Instead, the B molecules collide with A molecules in a variety of orientations and with a distribution of collision energies whose range depends on the Maxwell-Boltzmann distribution of kinetic energies of the B molecules in the bulb. The advantage of beam-bulb experiments is that one can achieve much higher collision densities than in crossed-beam experiments because the density of B molecules inside the bulb can be much higher than the densities achievable in a beam of B molecules.
There are cases in which the beam-bulb experiments can be used to determine how the reaction rate depends on collision energy even though the molecules in the bulb have a distribution of kinetic energies. That is, if the species in the beam have much higher kinetic energies than most of the B molecules, then the A + B collision energy is primarily determined by the beam energy. An example of this situation is provided by so-called guided-ion beam experiments in which a beam of ions having well-specified kinetic energy E impinges on molecules in a bulb having a temperature \(T\) for which \(kT \ll E\). Figure 8.6 illustrates data that can be extracted from such an experiment.
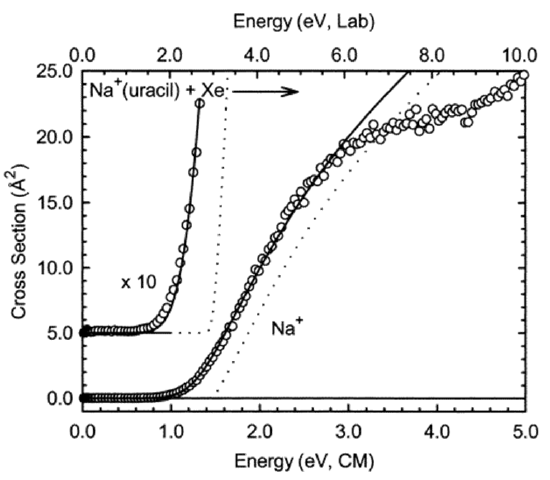
In Figure 8.6, we illustrate the cross-section \(\sigma\) (related to the bimolecular rate constant \(k\) by \(\sigma v = k\), where v is the relative collision speed) for production of \(Na^+\) ions when a beam of \(Na^+\)(uracil) complexes having energy E (the horizontal axis) collides with a bulb containing Xe atoms at room temperature. In this case, the reaction is simply the collision-induced dissociation (CID) process in which the complex undergoes unimolecular decomposition after gaining internal energy in collisions with Xe atoms:
\[Na^+({\rm uracil}) \rightarrow Na^+ + \rm uracil.\]
The primary knowledge gained in this CID experiment is the threshold energy \(E^*\); that is, the minimum collision energy needed to effect dissociation of the \(Na^+({\rm uracil})\) complex. This kind of data has proven to offer some of the most useful information about bond dissociation energies of a wide variety of species. In addition, the magnitude of the reaction cross-section \(\sigma\) as a function of collision energy is a valuable product of such experiments. These kind of CID beam-bulb experiments offer one of the most powerful and widely used means of determining such bond-rupture energies and reaction rate constants.
Other Methods
Of course, not all chemical reactions occur so quickly that they require the use of fast lasers to follow concentrations of reacting species or pump-probe techniques to generate and probe these molecules. For slower chemical reactions, one can use other methods for monitoring the relevant concentrations. These methods include electrochemistry (where the redox potential is the species’ signature) and NMR spectroscopy (where the chemical shifts of functional groups are the signatures) both of whose instrumental response times are too slow for probing fast reactions.
In addition, when the reactions under study do not proceed to completion but exist in equilibrium with a back reaction, alternative approaches can be used. The example discussed in Chapter 5 is one such case. Let us briefly review it here and again consider the reaction of an enzyme E and a substrate S to form the enzyme-substrate complex ES:
\[E + S \rightleftharpoons ES.\]
In the perturbation-type experiments, the equilibrium concentrations of the species are "shifted" by a small amount \(\delta\) by application of the perturbation, so that
\[[ES] = [ES]_{\rm eq} -\delta\]
\[[E] = [E]_{\rm eq} +\delta\]
\[[S] = [S]_{\rm eq} +\delta.\]
Subsequently, the following rate law will govern the time evolution of the concentration change d:
\[-\dfrac{d\delta}{dt} = - k_r ([ES]_{\rm eq} -\delta) + k_f ([E]_{\rm eq} +\delta) ([S]_{\rm eq} +\delta).\]
Assuming that \(\delta\) is very small (so that the term involving \(\delta^2\) cam be neglected) and using the fact that the forward and reverse rates balance at equilibrium, this equation for the time evolution of \(\delta\) can be reduced to:
\[-\dfrac{d\delta}{dt} = (k_r + k_f [S]_{\rm eq} + k_f[E]_{\rm eq}) \delta.\]
So, the concentration deviations from equilibrium will return to equilibrium exponentially with an effective rate coefficient that is equal to a sum of terms:
\[k_{\rm eff} = k_r + k_f [S]_{\rm eq} + k_f[E]_{\rm eq}.\]
So, by following the concentrations of the reactants or products as they return to their equilibrium values, one can extract the effective rate coefficient \(k_{\rm eff}\). Doing this at a variety of different initial equilibrium concentrations (e,g., \([S]_{\rm eq}\) and \([E]_{\rm eq}\)), and seeing how \(k_{\rm eff}\) changes, one can then determine both the forward and reverse rate constants.
Contributors and Attributions
Jack Simons (Henry Eyring Scientist and Professor of Chemistry, U. Utah) Telluride Schools on Theoretical Chemistry
Integrated by Tomoyuki Hayashi (UC Davis)