
9: Exercises
- Page ID
- 11559

The following are some problems that will help you refresh your memory about material you should have learned in undergraduate chemistry classes and that allow you to exercise the material taught in this text.
Chapters of Part 1
1.You should be able to set up and solve the one- and two-dimensional particle in a box Schrödinger equations. I suggest you now try this and make sure you see:
- How the second order differential equations have two independent solutions, so the most general solution is a sum of these two.
- How the two boundary conditions reduce the number of acceptable solutions from two to one and limit the values of \(E\) that can be “allowed”.
- How the wave function is continuous even at the box boundaries, but \(\dfrac{d\Psi}{dx}\) is not. In general \(\dfrac{d}{dx}\), which relates to the momentum because \(-i\hbar \dfrac{d}{dx}\) is the momentum operator, is continuous except at points where the potential \(V(x)\) undergoes an infinite jump as it does at the box boundaries. The infinite jump in \(V\), when viewed classically, means that the particle would undergo an instantaneous reversal in momentum at this point, so its momentum would not be continuous. Of course, in any realistic system, \(V\) does not have infinite jumps, so momentum will vary smoothly and thus \(\dfrac{d\Psi}{dx}\) will be continuous.
- How the energy levels grow with quantum number \(n\) as \(n^2\).
- What the wave functions look like when plotted.
2. You should go through the various wave functions treated in the Part 1 (e.g., particles in boxes, rigid rotor, harmonic oscillator) and make sure you see how the \(|\Psi|^2\) probability plots of such functions are not at all like the classical probability distributions except when the quantum number is very large.
3. You should make sure you understand how the time evolution of an eigenstate \(\Psi\) produces a simple \(\exp(-i tE/ \hbar)\) multiple of \(\Psi\) so that \(|\Psi|^2\) does not depend on time. However, when \(\Psi\) is not an eigenstate (e.g., when it is a combination of such states), its time propagation produces a \(\Psi\) whose \(|\Psi|^2\) probability distribution changes with time.
4. You should notice that the densities of states appropriate to the 1-, 2-, and 3- dimensional particle in a box problem (which relate to translations in these dimensions) depend of different powers of \(E\) for the different dimensions.
5. You should be able to solve 2x2 and 3x3 Hückel matrix eigenvalue problems both to obtain the orbital energies and the normalized eigenvectors. For practice, try to do so for
- the allyl radical’s three \(\pi\) orbitals
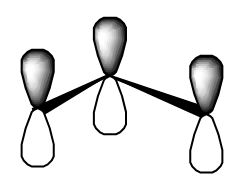
- the cyclopropenly radical’s three \(\pi\) orbitals.
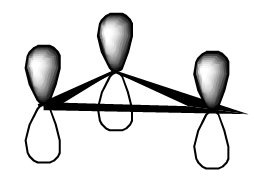
Do you see that the algebra needed to find the above sets of orbitals is exactly the same as was needed when we treat the linear and triangular sodium trimer?
6. You should be able to follow the derivation of the tunneling probability. Doing this offers a good test of your ability to apply the boundary conditions properly, so I suggest you do this task. You should appreciate how the tunneling probability decays exponentially with the “thickness” of the tunneling barrier and with the “height” of this barrier and that tunneling for heavier particles is less likely than for light particles. This is why tunneling usually is considered only for electrons, protons, and neutrons.
7. I do not expect that you could carry off a full solution to the Schrödinger equation for the hydrogenic atom. However, I think you need to pay attention to
- How separations of variables leads to a radial and two angular second order differential equations.
- How the boundary condition that and + 2 are equivalent points in space produces the m quantum number.
- How the l quantum number arises from the equation.
- How the condition that the radial wave function not “explode” (i.e., go to infinity) as the coordinate r becomes large gives rise to the equation for the energy \(E\).
- The fact that the angular parts of the wave functions are spherical harmonics, and that these are exactly the same wave functions for the rotational motion of a linear molecule.
- How the energy \(E\) depends on the \(n\) quantum number as \(n^{-2}\) and on the nuclear charge \(Z\) as \(Z^2\), and that the bound state energies are negative (do you understand what this means? That is, what is the zero or reference point of energy?).
8. You should make sure that you are familiar with how the rigid-rotor and harmonic oscillator energies vary with quantum numbers (\(J\), \(M\) in the former case, \(v\) in the latter). You should also know how these energies depend on the molecular geometry (in the former) and on the force constant and reduced mass (in the latter). You should note that \(E\) depends quadratically on \(J\) but linearly on \(v\).
9. You should know what the Morse potential is and what its parameters mean. You should understand that the Morse potential displays anharmonicity, but the harmonic potential does not.
10. You should be able to follow how the mass-weighted Hessian matrix can be used to approximate the vibrational motions of a polyatomic molecule. And, you should understand how the eigenvalues of this matrix produce the harmonic vibrational frequencies and the corresponding eigenvectors describe the motions of the molecule associated with these frequencies.
Practice with matrices and operators
1.Find the eigenvalues and corresponding normalized eigenvectors of the following matrices:
\[\left[\begin{array}{cc}
-1 & 2\\
2 & 2
\end{array}\right]\]
\[\left[\begin{array}{ccc}
-2 & 0 & 0\\
0 & -1 & 2 \\
0 & 2 & 2
\end{array}\right]\]
2. Replace the following classical mechanical expressions with their corresponding quantum mechanical operators:
- K.E. = \(\dfrac{mv^2}{2}\) in three-dimensional space.
- \(\textbf{p} = m\textbf{v}\), a three-dimensional Cartesian vector.
- \(y\)-component of angular momentum: \(L_y = zp_x - xp_z\).
3. Transform the following operators into the specified coordinates:
- \(L_x=\) from Cartesian to spherical polar coordinates.
- \(L_z=\) from spherical polar to Cartesian coordinates.
4. Match the eigenfunctions in column B to their operators in column A. What is the eigenvalue for each eigenfunction?
\[\begin{array}{lll}
\phantom{aaaaa} & \text{ Column A } & \text{ Column B } \\ \hline
{\rm i.} &(1-x^2) - x & 4x^4 - 12x^2 + 3 \\
{\rm ii.} & \dfrac{d^2}{dx^2} & 5x^4 \\
{\rm iii.}& x\dfrac{d}{dx} & e^{3x} + e^{-3x} \\
{\rm iv.}& \dfrac{d^2}{dx^2}- 2x\dfrac{d}{dx} & x^2 - 4x + 2 \\
{\rm v.}& x \dfrac{d^2}{dx^2}+ (1-x) \dfrac{d}{dx} & 4x^3 - 3x
\end{array}\]
Review of shapes of orbitals
5.Draw qualitative shapes of the (1) \(s\), (3) \(p\) and (5) \(d\) atomic orbitals (note that these orbitals represent only the angular portion and do not contain the radial portion of the hydrogen like atomic wave functions) Indicate with the relative signs of the wave functions and the position(s) (if any) of any nodes.
6.Plot the radial portions of the \(4s\), \(4p\), \(4d\), and \(4f\) hydrogen like atomic wave functions.
7. Plot the radial portions of the \(1s\), \(2s\), \(2p\), \(3s\), and \(3p\) hydrogen like atomic wave functions for the Si atom using screening concepts for any inner electrons.
Labeling orbitals using point group symmetry
8. Define the symmetry adapted "core" and "valence" atomic orbitals of the following systems:
- \(NH_3\) in the \(C_{3v}\) point group,
- \(H_2O\) in the \(C_{2v}\) point group,
- \(H_2O_2\) (cis) in the \(C_2\) point group
- \(N\) in \(D_{\infty h}\), \(D_{2h}\), \(C_{2v}\), and \(C_{s}\) point groups
- \(N_2\) in \(D_{\infty h}\), \(D_{2h}\), \(C_{2v}\), and \(C_{s}\) point groups.
problem to practice the basic tools of the Schrödinger equation
9. A particle of mass \(m\) moves in a one-dimensional box of length \(L\), with boundaries at \(x = 0\) and \(x = L\). Thus, \(V(x)\) = 0 for \(0 \le x \le L\), and \(V(x) = \infty\) elsewhere. The normalized eigenfunctions of the Hamiltonian for this system are given by \(\Psi_n(x) = \sqrt{\dfrac{2}{L}} \sin\dfrac{2\pi x}{L}\), with \(E_n =\dfrac{n^2\pi^2\hbar^2}{2mL^2}\), where the quantum number \(n\) can take on the values \(n=1,2,3,....\)
- Assuming that the particle is in an eigenstate, \(\Psi_n(x)\), calculate the probability that the particle is found somewhere in the region \(0 \le x \le \dfrac{L}{4}\). Show how this probability depends on \(n\).
- For what value of \(n\) is there the largest probability of finding the particle in \(0 \le x \le \dfrac{L}{4}\) ?
- Now assume that \(\Psi\) is a superposition of two eigenstates, \[\Psi = a\Psi_n + b\Psi_m, \text{ at time } t = 0. \] What is \(\Psi\) at time t? What energy expectation value does \(\Psi\) have at time t and how does this relate to its value at \(t = 0\)?
- For an experimental measurement which is capable of distinguishing systems in state \(\Psi_n\) from those in \(\Psi_m\), what fraction of a large number of systems each described by \(\Psi\) will be observed to be in \(\Psi_n\)? What energies will these experimental measurements find and with what probabilities?
- For those systems originally in \(\Psi = a\Psi_n + b\Psi_m\) which were observed to be in \(\Psi_n\) at time \(t\), what state (\(\Psi_n\), \(\Psi_m\), or whatever) will they be found in if a second experimental measurement is made at a time \(t'\) later than \(t\)?
- Suppose by some method (which need not concern us at this time) the system has been prepared in a nonstationary state (that is, it is not an eigenfunction of \(H\)). At the time of a measurement of the particle's energy, this state is specified by the normalized wave function \(\Psi = \sqrt{\dfrac{30}{L^5}}x(L-x)\) for \(0 \le x \le L\), and \(\Psi = 0\) elsewhere. What is the probability that a measurement of the energy of the particle will give the value \(E_n =\dfrac{n^2\pi^2\hbar^2}{2mL^2}\) for any given value of \(n\)?
- What is the expectation value of \(H\), i.e. the average energy of the system, for the wave function \(\Psi\) given in part f?
problem on the properties of non-stationary states
10. Show that for a system in a non-stationary state,
\(\Psi = \sum_j C_j\Psi_j e^{-iE_jt/\hbar}\), the average value of the energy does not vary with time but the expectation values of other properties do vary with time.
problem about Jahn-Teller distortion
11. The energy states and wave functions for a particle in a 3-dimensional box whose lengths are \(L_1\), \(L_2\), and \(L_3\) are given by
\[E(n_1,n_2,n_3) = \dfrac{h^2}{8m}\left[\Big(\dfrac{n_1}{L_1}\Big)^2+\Big(\dfrac{n_2}{L_2}\Big)^2+\Big(\dfrac{n_3}{L_3}\Big)^2\right]\text{ and}\]
\[\Psi(n_1,n_2,n_3) = \sqrt{\dfrac{2}{L_1}}\sqrt{\dfrac{2}{L_2}}\sqrt{\dfrac{2}{L_3}} \sin\Big(\dfrac{n_1\pi x}{L_1}\Big) \sin\Big(\dfrac{n_2\pi x}{L_2}\Big) \sin\Big(\dfrac{n_3\pi x}{L_3}\Big).\]
These wave functions and energy levels are sometimes used to model the motion of electrons in a central metal atom (or ion) which is surrounded by six ligands in an octahedral manner.
- Show that the lowest energy level is nondegenerate and the second energy level is triply degenerate if \(L_1= L_2= L_3\). What values of \(n_1\), \(n_2\), and \(n_3\) characterize the states belonging to the triply degenerate level?
- For a box of volume \(V = L_1L_2L_3\) show that for three electrons in the box (two in the nondegenerate lowest "orbital", and one in the next), a lower total energy will result if the box undergoes a rectangular distortion (\(L_1= L_2\ne L_3\)). which preserves the total volume than if the box remains undistorted (hint: if \(V\) is fixed and \(L_1 = L_2\), then \(L_3 =\dfrac{V}{L_1^2}\) and \(L_1\) is the only "variable").
- Show that the degree of distortion (ratio of \(L_3\) to \(L_1\)) which will minimize the total energy is \(L_3 = \sqrt{2}L_1\). How does this problem relate to Jahn-Teller distortions? Why (in terms of the property of the central atom or ion) do we do the calculation with fixed volume?
- By how much (in eV) will distortion lower the energy (from its value for a cube, \(L_1= L_2= L_3\)) if \(V\) = 8 Å and = 6.01 x 10 erg cm2. 1 eV = 1.6 x 10 erg
particle on a ring model for electrons moving in cyclic compounds
12. The \(\pi\)-orbitals of benzene, \(C_6H_6\), may be modeled very crudely using the wave functions and energies of a particle on a ring. Lets first treat the particle on a ring problem and then extend it to the benzene system.
- Suppose that a particle of mass m is constrained to move on a circle (of radius \(r\)) in the \(xy\) plane. Further assume that the particle's potential energy is constant (choose zero as this value). Write down the Schrödinger equation in the normal Cartesian coordinate representation. Transform this Schrödinger equation to cylindrical coordinates where \(x = r\cos\theta\), \(y = r\sin\phi\), and \(z = z\) (\(z = 0\) in this case). Taking \(r\) to be held constant, write down the general solution, \(\Phi(\phi)\), to this Schrödinger equation. The "boundary" conditions for this problem require that \(\Phi(\phi) = \Phi(\phi+2\pi)\). Apply this boundary condition to the general solution. This results in the quantization of the energy levels of this system. Write down the final expression for the normalized wave functions and quantized energies. What is the physical significance of these quantum numbers that can have both positive and negative values? Draw an energy diagram representing the first five energy levels.
- Treat the six \(\pi\)-electrons of benzene as particles free to move on a ring of radius 1.40 Å, and calculate the energy of the lowest electronic transition. Make sure the Pauli principle is satisfied! What wavelength does this transition correspond to? Suggest some reasons why this differs from the wavelength of the lowest observed transition in benzene, which is 2600 Å.
non-stationary state wave function
13. A diatomic molecule constrained to rotate on a flat surface can be modeled as a planar rigid rotor (with eigenfunctions, \(\Phi(\phi)\), analogous to those of the particle on a ring of problem 12) with fixed bond length \(r\). At \(t = 0\), the rotational (orientational) probability distribution is observed to be described by a wave function \(\Psi(\phi,0) = \sqrt{\dfrac{4}{3\pi}}\cos^2\phi\). What values, and with what probabilities, of the rotational angular momentum, \(-i\hbar\dfrac{\partial}{\partial \phi}\), could be observed in this system? Explain whether these probabilities would be time dependent as \(\Psi(\phi,0)\) evolves into \(\Psi(\phi,t)\).
problem about Franck-Condon factors
14. Consider an \(N_2\) molecule, in the ground vibrational level of the ground electronic state, which is bombarded by 100 eV electrons. This leads to ionization of the \(N_2\) molecule to form \(N\). In this problem we will attempt to calculate the vibrational distribution of the newly-formed \(N\) ions, using a somewhat simplified approach.
- Calculate (according to classical mechanics) the velocity (in cm/sec) of a 100 eV electron, ignoring any relativistic effects. Also calculate the amount of time required for a 100 eV electron to pass an Nmolecule, which you may estimate as having a length of 2Å.
- The radial Schrödinger equation for a diatomic molecule treating vibration as a harmonic oscillator can be written as:
\[ -\dfrac{\hbar^2}{2\mu r^2}\left(\dfrac{\partial}{\partial r}\left( r^2 \dfrac{\partial \Psi}{\partial r}\right)\right)+ \dfrac{k}{2}(r-r_e)^2 \Psi =E\Psi,\]
Substituting \(\Psi(r) =\dfrac{F(r)}{r} \), this equation can be rewritten as:
\[-\dfrac{\hbar^2}{2\mu r^2}\dfrac{\partial^2}{\partial r^2}F(r) + \dfrac{k}{2}(r-r_e)^2F(r) = EF(r) .\]
The vibrational Hamiltonian for the ground electronic state of the \(N_2\) molecule within this approximation is given by:
\[H({\rm N_2}) = \dfrac{\hbar^2}{2\mu r^2}\dfrac{\partial^2}{\partial r^2}+ \dfrac{k_{\rm N_2}}{2}(r-r_{\rm N_2})^2,\]
where \(r_{\rm N_2}\) and \(k_{\rm N_2}\) have been measured experimentally to be:
\[ r_{\rm N_2} = 1.09769 Å; \hspace{1cm} k_{\rm N_2} = 2.294 \times 10^6 \dfrac{\rm g}{\rm sec^2}.\]
The vibrational Hamiltonian for the \(N_2^+\) ion, however, is given by :
\[H({\rm N_2^+}) = \dfrac{\hbar^2}{2\mu r^2}\dfrac{\partial^2}{\partial r^2}+ \dfrac{k_{\rm N_2^+}}{2}(r-r_{\rm N_2^+})^2,\]
where \(r_{\rm N_2^+}\) and \(k_{\rm N_2^+}\) have been measured experimentally to be:
\[r_{\rm N_2^+}= 1.11642 Å;\hspace{1cm} k_{\rm N_2} = 2.009 \times 10^6 \dfrac{\rm g}{\rm sec^2}.\]
In both systems the reduced mass is \(\mu = 1.1624 \times 10^{-23}\) g. Use the above information to write out the ground state vibrational wave functions of the \(N_2\) and \(N\) molecules, giving explicit values for any constants which appear in them. The \(v = 0\) harmonic oscillator function is \(\Psi_0 = \Big(\dfrac{\alpha}{\pi}\Big)^{1/4} \exp(-\alpha x^2/2)\).
c. During the time scale of the ionization event (which you calculated in part a), the vibrational wave function of the \(N_2\) molecule has effectively no time to change. As a result, the newly-formed \(N\) ion finds itself in a vibrational state which is not an eigenfunction of the new vibrational Hamiltonian, \(H({\rm N}_2^+)\). Assuming that the \(N_2\) molecule was originally in its \(v=0\) vibrational state, calculate the probability that the \(N\) ion will be produced in its \(v=0\) vibrational state.
Vibration of a diatomic molecule
15.The force constant, \(k\), of the \(C-O\) bond in carbon monoxide is 1.87 x 10 g/sec. Assume that the vibrational motion of \(CO\) is purely harmonic and use the reduced mass \(\mu= 6.857\) amu.
Calculate the spacing between vibrational energy levels in this molecule, in units of ergs and cm-1.
Calculate the uncertainty in the internuclear distance in this molecule, assuming it is in its ground vibrational level. Use the ground state vibrational wave function (\(\Psi_{v=0}\); recall that I gave you this function in problem 14), and calculate \(\langle x\rangle \), \(\langle x^2\rangle \), and \(\Delta x = \sqrt{\langle x^2\rangle - \langle x\rangle ^2}\).
Under what circumstances (i.e. large or small values of \(k\); large or small values of \(\mu\)) is the uncertainty in internuclear distance large? Can you think of any relationship between this observation and the fact that helium remains a liquid down to absolute zero?
Variational Method Problem
16. A particle of mass m moves in a one-dimensional potential whose Hamiltonian is given by
\[H = -\dfrac{\hbar^2}{2m}\dfrac{d^2}{dx^2}+ a|x| ,\]
where the absolute value function is defined by \(|x| = x\) if \(x \ge 0\) and \(|x| = -x\) if \(x \le 0\).
- Use the normalized trial wavefunction \(\phi = \Big(\dfrac{2b}{\pi}\Big)^{\frac{1}{4}}e^{-bx^2}\) to estimate the energy of the ground state of this system, using the variational principle to evaluate \(W(b)\), the variational expectation value of \(H\).
- Optimize b to obtain the best approximation to the ground state energy of this system, using a trial function of the form of \(\phi\), as given above. The numerically calculated exact ground state energy is \(0.808616 \hbar^{\frac{2}{3}}m^{-\frac{1}{3}}a^{-\frac{2}{3}}\). What is the percent error in your value?
Another Variational Method Problem
17. The harmonic oscillator is specified by the Hamiltonian:
\[H = -\dfrac{\hbar^2}{2m}\dfrac{d^2}{dx^2}+ \dfrac{1}{2}kx^2 ,\]
Suppose the ground state solution to this problem were unknown, and that you wish to approximate it using the variational theorem. Choose as your trial wavefunction,
\[\begin{array}{ll}
\phi = \sqrt{\dfrac{15}{16}}a^{-\frac{5}{2}} & \text{for } -a < x < a\\
\phi = 0 & \text{for } |x|\ge a
\end{array}\]
where a is an arbitrary parameter which specifies the range of the wavefunction. Note that f is properly normalized as given.
a. Calculate \(\displaystyle\int_{-\infty}^{\infty}\phi^*H\phi\, dx\) and show it to be given by:
\[\int_{-\infty}^{\infty}\phi^*H\phi\, dx=\dfrac{5}{4}\dfrac{\hbar^2}{ma} + \dfrac{ka^2}{14}.\]
b. Calculate \(\displaystyle\int_{-\infty}^{\infty}\phi^*H\phi\, dx\) for \(a = b\Big(\dfrac{\hbar^2}{km}\Big)^{\frac{1}{4}}\).
c. To find the best approximation to the true wavefunction and its energy, find the minimum of \(\displaystyle\int_{-\infty}^{\infty}\phi^*H\phi\, dx\) by setting \(\displaystyle\dfrac{d}{da}\int_{-\infty}^{\infty}\phi^*H\phi\, dx= 0\) and solving for \(a\). Substitute this value into the expression for \(\displaystyle\int_{-\infty}^{\infty}\phi^*H\phi\, dx\) given in part a. to obtain the best approximation for the energy of the ground state of the harmonic oscillator.
d. What is the percent error in your calculated energy of part c.?
Perturbation Theory Problem
18. Consider an electron constrained to move on the surface of a sphere of radius \(r_0\). The Hamiltonian for such motion consists of a kinetic energy term only \(H_0 = \dfrac{L^2}{2m_er_0^2}\), where \(L\) is the orbital angular momentum operator involving derivatives with respect to the spherical polar coordinates (\(\theta,\phi\)). \(H_0\) has the complete set of eigenfunctions \(\psi= Y_{l,m}(\theta,\phi)\).
a. Compute the zeroth order energy levels of this system.
b. A uniform electric field is applied along the \(z\)-axis, introducing a perturbation \(V = -e\varepsilon z = -e\varepsilon r_0\cos\theta\), where \(\varepsilon\) is the strength of the field. Evaluate the correction to the energy of the lowest level through second order in perturbation theory, using the identity
\[ \cos\theta Y_{l,m}(\theta,\phi) = \sqrt{\dfrac{(l+m+1)(l-m+1)}{(2l+1)(2l+3)}}Y_{l+1,m}(\theta,\phi) + \sqrt{\dfrac{(l+m)(l-m)}{(2l+1)(2l-1)}}Y_{l-1,m}(\theta,\phi) .\]
Note that this identity enables you to utilize the orthonormality of the spherical harmonics.
c. The electric polarizability \(\alpha\) gives the response of a molecule to an externally applied electric field, and is defined by \(\alpha = -\dfrac{\partial^2 E}{\partial \varepsilon^2}\) where \(E\) is the energy in the presence of the field and \(\varepsilon\) is the strength of the field. Calculate \(\alpha\) for this system.
d. Use this problem as a model to estimate the polarizability of a hydrogen atom, where \(r_0 = a_0 = 0.529\) Å, and a cesium atom, which has a single 6s electron with \(r_0 \approx 2.60 Å\). The corresponding experimental values are \(\alpha_H = 0.6668 Å^3\) and \(\alpha_{Cs} = 59.6 Å^3\).
Hartree-Fock problem you can do by hand
19. Given the following orbital energies (in hartrees) for the \(N\) atom and the coupling elements between two like atoms (these coupling elements are the Fock matrix elements from standard ab-initio minimum-basis SCF calculations), calculate the molecular orbital energy levels and orbitals. Draw the orbital correlation diagram for formation of the \(N_2\) molecule. Indicate the symmetry of each atomic and each molecular orbital. Designate each of the molecular orbitals as bonding, non-bonding, or antibonding.
\[N_{1s} = -15.31^*\]
\[ N_{2s} = -0.86^*\]
\[N_{2p} = -0.48^*\]
\(N_2\) \(\sigma_g\) Fock matrix*
\[\left[\begin{array}{ccc}
-6.52 & & \\
-6.22 & -7.06 & \\
3.61 & 4.00 & -3.92
\end{array}\right]\]
\(N_2\) \(\pi_g\) Fock matrix*
\[[0.28]\]
\(N_2\) \(\sigma_u\) Fock matrix*
\[\left[\begin{array}{ccc}
1.02 & & \\
-0.60 & -7.59 & \\
0.02 & 7.42 & -8.53
\end{array}\right]\]
\(N_2\) \(\pi_u\) Fock matrix*
\[-0.58\]
*The Fock matrices (and orbital energies) were generated using standard minimum basis set SCF calculations. The Fock matrices are in the orthogonal basis formed from these orbitals.
orbital correlation diagram problem
20. Given the following valence orbital energies for the \(C\) atom and \(H_2\) molecule, draw the orbital correlation diagram for formation of the \(CH_2\) molecule (via a \(C_{2v}\) insertion of \(C\) into \(H_2\) resulting in bent \(CH_2\)). Designate the symmetry of each atomic and molecular orbital in both their highest point group symmetry and in that of the reaction path (\(C_{2v}\)).
\[\begin{array}{ll}
C_{1s} = -10.91^* & H_2\; \sigma_g = -0.58^*\\
C_{2s} = -0.60^* & H_2\; \sigma_u = 0.67^*\\
C_{2p} = -0.33^* & \end{array}\]
*The orbital energies were generated using standard STO3G minimum basis set SCF calculations.
Practice using point group symmetry
21. Qualitatively analyze the electronic structure (orbital energies and orbitals) of \(PF_5\). Analyze only the \(3s\) and \(3p\) electrons of P and the one \(2p\) bonding electron of each \(F\). Proceed with a \(D_{3h}\) analysis in the following manner:
- Symmetry adapt the top and bottom \(F\) atomic orbitals.
- Symmetry adapt the three (trigonal) \(F\) atomic orbitals.
- Symmetry adapt the P \(3s\) and \(3p\) atomic orbitals.
- Allow these three sets of \(D_{3h}\) orbitals to interact and draw the resultant orbital energy diagram.
- Symmetry label each of these molecular energy levels. Fill this energy diagram with 10 "valence" electrons.
Practice with term symbols and determinental wave functions for atoms and molecules
22. For the given orbital occupations (configurations) of the following systems, determine all possible states (all possible allowed combinations of spin and space states). There is no need to form the determinental wave functions, simply label each state with its proper term symbol.
\[\begin{array}{lll}
{\rm a.} & CH_2 & 1a_1{}^2\,2a_1{}^2\,1b_2{}^2\,3a_1{}^1\,1b_1{}^1\\
{\rm b.} & B_2 & 1\sigma_g{}^2\,1\sigma_u{}^2\,2\sigma_g{}^2\,2\sigma_u{}^2\,1\pi_u{}^1\,2\pi_u{}^1\\
{\rm c.} & O_2 & 1\sigma_g{}^2\,1\sigma_u{}^2\,2\sigma_g{}^2\,2\sigma_u{}^2\,1\pi_u{}^4\,3\sigma_g{}^2\,1\pi_g{}^2 \\
{\rm d.} & Ti & 1s{}^2\,2s{}^2\,2p{}^6\,3s{}^2\,3p{}^6\,4s{}^2\,3d{}^1\,4d{}^1\\
{\rm e.} & Ti & 1s{}^2\,2s{}^2\,2p{}^6\,3s{}^2\,3p{}^6\,4s{}^2\,3d{}^2
\end{array}\]
23. Construct Slater determinant wave functions for each of the following states of \(CH_2\):
- \(^1B_1\) (\(1a_1{}^2\,2a_1{}^2\,1b_2{}^2\,3a_1{}^1\,1b_1{}^1\))
- \(^3B_1\) (\(1a_1{}^2\,2a_1{}^2\,1b_2{}^2\,3a_1{}^1\,1b_1{}^1\))
- \(^1A_1\) (\(1a_1{}^2\,2a_1{}^2\,1b_2{}^2\,3a_1{}^2\))
Woodward-Hoffmann rules problem
24. Let us investigate the reactions:
i. \(CH_2(^1A_1) \rightarrow H_2 + C\), and
ii. \(CH_2(^3B_1) \rightarrow H_2 + C\),
under an assumed \(C_{2v}\) reaction pathway utilizing the following information:
\(C\) atom: \(^3P\) \(^1D\) \(^1S\)
\(C(^3P) + H_2 \rightarrow CH_2(^3B_1)\) \(\Delta E = -78.8\) kcal/mole
\(C(^1D) + H_2 \rightarrow CH_2(^1A_1)\) \(\Delta E = -97.0\) kcal/mole
IP (\(H_2\)) > IP (2s carbon).
- Write down (first in terms of \(2p_{1,0,-1}\) orbitals and then in terms of \(2p_{x,y,z}\) orbitals) the:
- three Slater determinant (SD) wave functions belonging to the \(^3P\) state all of which have \(M_S = 1\),
- five \(^1D\) SD wave functions, and
- one \(^1S\) SD wave function.
- Using the coordinate system shown below, label the hydrogen orbitals sg, su and the carbon \(2s\), \(2p_x\), \(2p_y\), \(2p_z\), orbitals as \(a_1, b_1(x), b_(y),\) or \(a_2\). Do the same for the \(\sigma\), \(\sigma\), \(\sigma^*\), \(\sigma^*\), \(n\), and \(\pi_p\) orbitals of \(CH_2\).
- Draw an orbital correlation diagram for the \(CH_2 \rightarrow H_2 + C\) reactions. Try to represent the relative energy orderings of the orbitals correctly.
- Draw a configuration correlation diagram for \(CH_2(^3B_1) \rightarrow H_2 + C\) showing all configurations which arise from the \(C(^3P) + H_2\) products. You can assume that doubly excited configurations lie much (~100 kcal/mole) above their parent configurations.
- Repeat step d. for \(CH_2(^1A_1) \rightarrow H_2 + C\) again showing all configurations which arise from the \(C(^1D) + H_2 \) products.
- Do you expect the reaction \(C(^3P) + H_2 \rightarrow CH_2\) to have a large activation barrier? About how large? What state of \(CH_2\) is produced in this reaction? Would distortions away from \(C_{2v}\) symmetry be expected to raise or lower the activation barrier? Show how one could estimate where along the reaction path the barrier top occurs.
- Would \(C(^1D) + H_2 \rightarrow CH_2\) be expected to have a larger or smaller barrier than you found for the \(^3P\) \(C\) reaction?
Another Woodward-Hoffmann rule problem
25. The decomposition of the ground-state singlet carbene,

to produce acetylene and \(^1D\) carbon is known to occur with an activation energy equal to the reaction endothermicity. However, when the corresponding triplet carbene decomposes to acetylene and ground-state (triplet) carbon, the activation energy exceeds this reaction's endothermicity. Construct orbital, configuration, and state correlation diagrams that permit you to explain the above observations. Indicate whether single configuration or configuration interaction wave functions would be required to describe the above singlet and triplet decomposition processes.
Practice with rotational spectrocopy and its relation to molecular structure
26. Consider the molecules \(CCl_4\), \(CHCl_3\), and \(CH_2Cl_2\).
- What kind of rotor are they (symmetric top, etc; do not bother with oblate, or near-prolate, etc.)
- Will they show pure rotational (i.e., microwave) spectra?
27. Assume that ammonia shows a pure rotational spectrum. If the rotational constants are 9.44 and 6.20 cm-1, use the energy expression:
\[E = (A - B) K^2 + B J(J + 1),\]
to calculate the energies (in cm-1) of the first three lines (i.e., those with lowest \(K\), \(J\) quantum number for the absorbing level) in the absorption spectrum (ignoring higher order terms in the energy expression).
problem on vibration-rotation spectroscopy
28. The molecule \(^{11}B ^{16}O\) has a vibrational frequency \(\omega_e = 1885\) cm-1, a rotational constant \(B_e = 1.78\) cm-1, and a bond energy from the bottom of the potential well of \(D= 8.28\) eV. Use integral atomic masses in the following:
In the approximation that the molecule can be represented as a Morse oscillator, calculate the bond length, \(R_e\) in angstroms, the centrifugal distortion constant, \(D_e\) in cm-1, the anharmonicity constant, \(\omega_e x_e\) in cm-1, the zero-point corrected bond energy, \(D_0^0\) in eV, the vibration rotation interaction constant, \(\alpha_e\) in cm-1, and the vibrational state specific rotation constants, \(B_0\) and \(B_1\) in cm-1. Use the vibration-rotation energy expression for a Morse oscillator:
\[E = \hbar\omega_e\Big(v + \dfrac{1}{2}\Big) - \hbar\omega_e x_e\Big(v + \dfrac{1}{2}\Big)^2 + B_vJ(J + 1) - D_eJ^2(J + 1)^2,\] where
\[Bv = B_e - \alpha_e\Big(v + \dfrac{1}{2}\Big),\; \alpha_e = \dfrac{-6B_e^2}{\hbar\omega_e}+ \dfrac{6\sqrt{B_e^3\hbar\omega_e x_e}}{\hbar\omega}, \; \text{and } D_e = \dfrac{4B_e^3}{\hbar\omega^2}.\]
Will this molecule show a pure rotation spectrum? A vibration-rotation spectrum? Assuming that it does, what are the energies (in cm-1) of the first three lines in the P branch (\(\Delta v = +1, \Delta J = -1\)) of the fundamental absorption?
problem labeling vibrational modes by symmetry
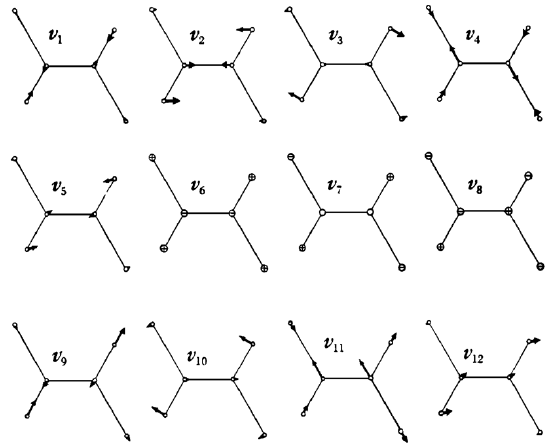
29. Consider trans-\(C_2H_2Cl_2\). The vibrational normal modes of this molecule are shown
below. What is the symmetry of the molecule? Label each of the modes with the appropriate irreducible representation.
problem in rotational spectroscopy
30. Suppose you are given two molecules (one is \(CH_2\) and the other is \(CH_2^-\) but you don't know which is which). Both molecules have \(C_{2v}\) symmetry. The \(CH\) bond length of molecule I is 1.121 Å and for molecule II it is 1.076 Å. The bond angle of molecule I is 104° and for molecule II it is 136°.
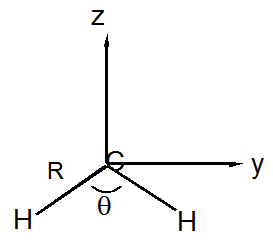
a. Using a coordinate system centered on the \(C\) nucleus as shown above (the molecule is in the \(YZ\) plane), compute the moment of inertia tensors of both species (I and II). The definitions of the components of the tensor are, for example:
\[I_{xx} = - \sum_j m_j (y_j^2+z_j^2) - M(Y^2 + Z^2)\]
\[I_{xy} = -\sum_j m_j x_j y_j - MXY\]
Here, \(m_j\) is the mass of the nucleus \(j\), \(M\) is the mass of the entire molecule, and \(X\), \(Y\), \(Z\) are the coordinates of the center of mass of the molecule. Use Å for distances and amu's for masses.
b. Find the principal moments of inertia \(I_a < I_b < I_c\) for both compounds ( in amu Å2 units) and convert these values into rotational constants \(A\), \(B\), and \(C\) in cm-1 using, for example,
\[A = \dfrac{h}{8\pi^2cI_a}.\]
c. Both compounds are "nearly prolate tops" whose energy levels can be well approximated using the prolate top formula:
\[E = (A - B) K^2 + BJ(J + 1),\]
if one uses for the \(B\) constant the average of the \(B\) and \(C\) valued determined earlier. Thus, take the \(B\) and \(C\) values (for each compound) and average them to produce an effective \(B\) constant to use in the above energy formula. Write down (in cm-1 units) the energy formula for both species. What values are \(J\) and \(K\) allowed to assume? What is the degeneracy of the level labeled by a given \(J\) and \(K\)?
d. Draw a picture of both compounds and show the directions of the three principle axes (a,b,c). On these pictures, show the kind of rotational motion associated with the quantum number \(K\).
e. Suppose you are given the photoelectron spectrum of \(CH_2^-\). In this spectrum \(J_j = J_i + 1\) transitions are called R-branch absorptions and those obeying \(J_j = J_i - 1\) are called P-branch transitions. The spacing between lines can increase or decrease as functions of \(J_i\) depending on the changes in the moment of inertia for the transition. If spacings grow closer and closer, we say that the spectrum exhibits a so-called band head formation. In the photoelectron spectrum that you are given, a rotational analysis of the vibrational lines in this spectrum is carried out and it is found that the R-branches show band head formation but the P-branches do not. Based on this information, determine which compound I or II is the \(CH_2^-\) anion. Explain you reasoning.
f. At what \(J\) value (of the absorbing species) does the band head occur and at what rotational energy difference?
Using point group symmetry in vibrational spectroscopy
31. Let us consider the vibrational motions of benzene. To consider all of the vibrational modes of benzene we should attach a set of displacement vectors in the \(x\), \(y\), and \(z\) directions to each atom in the molecule (giving 36 vectors in all), and evaluate how these transform under the symmetry operations of \(D_{6h}\). For this problem, however, let's only inquire about the \(C-H\) stretching vibrations.
a. Represent the \(C-H\) stretching motion on each \(C-H\) bond by an outward-directed vector on each \(H\) atom, designated \(r_i\):
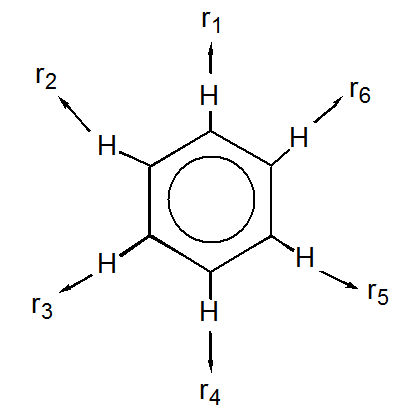
b. These vectors form the basis for a reducible representation. Evaluate the characters for this reducible representation under the symmetry operations of the \(D_{6h}\) group.
c. Decompose the reducible representation you obtained in part b. into its irreducible components. These are the symmetries of the various \(C-H\) stretching vibrational modes in benzene.
d. The vibrational state with zero quanta in each of the vibrational modes (the ground vibrational state) of any molecule always belongs to the totally symmetric representation. For benzene, the ground vibrational state is therefore of \(A_{1g}\) symmetry. An excited state which has one quantum of vibrational excitation in a mode which is of a given symmetry species has the same symmetry species as the mode which is excited (because the vibrational wave functions are given as Hermite polynomials in the stretching coordinate). Thus, for example, excitation (by one quantum) of a vibrational mode of \(A_{2u}\) symmetry gives a wave function of \(A_{2u}\) symmetry. To resolve the question of what vibrational modes may be excited by the absorption of infrared radiation we must examine the \(x\), \(y\), and \(z\) components of the transition dipole integral for initial and final state wave functions \(\psi_i\) and \(\psi_f\), respectively:
\[|\langle\psi_f | x |\psi_i \rangle| ,\;\; |\langle\psi_f | y |\psi_i \rangle| ,\; \; \text{and } |\langle\psi_f | z |\psi_i \rangle| .\]
Using the information provided above, which of the \(C-H\) vibrational modes of benzene will be infrared-active, and how will the transitions be polarized? How many \(C-H\) vibrations will you observe in the infrared spectrum of benzene?
e. A vibrational mode will be active in Raman spectroscopy only if one or more of the following integrals is nonzero:
\[|\langle\psi_f | xy |\psi_i \rangle| ,\;\;|\langle\psi_f | xz |\psi_i \rangle| , \;\; |\langle\psi_f | yz |\psi_i \rangle| ,\]
\[|\langle\psi_f | x^2 |\psi_i \rangle| ,\;\; |\langle\psi_f | y^2 |\psi_i \rangle| ,\;\; \text{and } |\langle\psi_f | z^2 |\psi_i \rangle| .\]
Using the fact that the quadratic operators transform according to the irreducible representations:
\[(x^2 + y^2, z^2) \Rightarrow A_{1g}\]
\[(xz, yz) \Rightarrow E_{1g}\]
\[(x^2 - y^2, xy) \Rightarrow E_{2g}\]
Determine which of the \(C-H\) vibrational modes will be Raman-active.
f. Are there any of the \(C-H\) stretching vibrational motions of benzene which cannot be observed in either infrared of Raman spectroscopy? Give the irreducible representation label for these unobservable modes.
problem on electronic spectra and lifetimes
32. Time dependent perturbation theory provides an expression for the radiative lifetime of an excited electronic state, given by \(t_R\):
\[t_R = \dfrac{3\hbar^4c^3}{4(E_i-E_f)^3|\mu_{fi}|^2},\]
where \(i\) refers to the excited state, \(f\) refers to the lower state, and \(\mu_{fi}\) is the transition dipole.
a. Evaluate the \(z\)-component of the transition dipole for the \(2p_z \rightarrow 1s\) transition in a hydrogenic atom of nuclear charge \(Z\), given:
\[\psi_{1s} =\dfrac{1}{\sqrt{\pi}} \bigg(\dfrac{Z}{a_0}\bigg)^{\frac{3}{2}} \exp\bigg(-\dfrac{Zr}{a_0}\bigg) ,\text{ and } \psi_{2p_z} =\dfrac{1}{4\sqrt{2\pi}} \bigg(\dfrac{Z}{a_0}\bigg)^{\frac{5}{2}} r \cos\theta\exp\bigg(-\dfrac{Zr}{2a_0}\bigg).\]
Express your answer in units of \(ea_0\).
b. Use symmetry to demonstrate that the \(x-\) and \(y\)-components of \(\mu_{fi}\) are zero, i.e.
\[\langle 2p_z| e x |1s\rangle = \langle 2p_z| e y |1s\rangle = 0.\]
c. Calculate the radiative lifetime \(t_R\) of a hydrogenlike atom in its \(2p_z\) state. Use the relation \(e^2 =\dfrac{\hbar^2}{m_ea_0}\) to simplify your results.
difference between slowly and quickly turning on a perturbation
33. Consider a case in which the complete set of states {\(\phi_k\)} for a Hamiltonian is known.
a. If the system is initially in the state m at time \(t=0\) when a constant perturbation \(V\) is suddenly turned on, find the probability amplitudes \(C_k^{(2)}(t)\) and \(C_m^{(2)}(t)\), to second order in \(V\), that describe the system being in a different state \(k\) or the same state \(m\) at time \(t\).
b. If the perturbation is turned on adiabatically (i.e., very slowly), what are \(C_k^{(2)}(t)\) and \(C_m^{(2)}(t)\)? Here, consider that the initial time is \(t_0 \rightarrow -\infty\), and the potential is \(V e^\eta t\), where the positive parameter \(\eta\) is allowed to approach zero \(\eta\rightarrow 0\) in order to describe the adiabatically turned on perturbation.
c. Compare the results of parts a. and b. and explain any differences.
d. Ignore first order contributions (assume they vanish) and evaluate the transition rates
\(|C_k^{(2)}(t)|^2\) for the results of part b. by taking the limit \(\eta \rightarrow 0^+\), to obtain the adiabatic results.
example of quickly turning on a perturbation- the sudden approximation
34. Consider an interaction or perturbation which is carried out suddenly (instantaneously, e.g., within an interval of time \(\Delta t\) which is small compared to the natural period \(\omega_{nm}^{-1}\) corresponding to the transition from state \(m\) to state \(n\)), and after that is turned off adiabatically (i.e., extremely slowly as \(V e^\eta t\)). The transition probability in this case is given as:
\[T_{nm} \approx \dfrac{|\langle n|V|m \rangle|^2}{\hbar^2\omega_{nm}^2}\]
where \(V\) corresponds to the maximum value of the interaction when it is turned on. This formula allows one to calculate the transition probabilities under the action of sudden perturbations which are small in absolute value whenever perturbation theory is applicable.
Let's use this "sudden approximation" to calculate the probability of excitation of an electron under a sudden change of the charge of the nucleus. Consider the reaction:
\[^3_1H \rightarrow ^3_2He^+ + e^-,\]
and assume the tritium atom has its electron initially in a \(1s\) orbital.
a. Calculate the transition probability for the transition \(1s \rightarrow 2s\) for this reaction using the above formula for the transition probability.
b. Suppose that at time \(t = 0\) the system is in a state which corresponds to the
wave function \(\phi_m\), which is an eigenfunction of the operator \(H_0\). At \(t = 0\), the sudden change of the Hamiltonian occurs (now denoted as \(H\) and remains unchanged). Calculate the same \(1s \rightarrow 2s\) transition probability as in part a., only this time as the square of the magnitude of the coefficient, \(A_{1s,2s}\) using the expansion:
\[\Psi(r,0) =\phi_m(r) =\sum_n A_{nm}\psi_n(r) ,\] where \[A_{nm} =\int \phi_m(r)\psi_n(r)d^3r\]
Note, that the eigenfunctions of \(H\) are \(\psi_n\) with eigenvalues \(E_n\). Compare this value with that obtained by perturbation theory in part a.
symmetric top rotational spectrum problem
35. The methyl iodide molecule is studied using microwave (pure rotational)
spectroscopy. The following integral governs the rotational selection rules for transitions labeled \(J, M, K \rightarrow J', M', K'\):
\[I = <D_{M'K'}^{J'}|\boldsymbol{\varepsilon}\bullet\boldsymbol{\mu}|D_{MK}^J>.\]
The dipole moment \(\boldsymbol{\mu}\) lies along the molecule's \(C_3\) symmetry axis. Let the electric field of the light \(\boldsymbol{\mu}\) define the lab-fixed Z-direction.
a. Using the fact that \(\cos\beta = D_{00}^*\), show that
\[I = 8\pi^2\mu\varepsilon(-1)^{(M+K)} \left(\begin{array}{ccc}J' & 1 & J \\ M & 0 & M \end{array}\right) \left(\begin{array}{ccc}J' & 1 & J \\ K & 0 & K \end{array}\right) \delta_{M'M}\delta_{K'K}\]
b. What restrictions does this result place on \(\Delta J = J' - J\)? Explain physically why the \(K\) quantum number can not change.
problem in electronic and photo-electron spectroscopy
36. Consider the molecule \(BO\).
a. What are the total number of possible electronic states that can be formed by combination of ground-state \(B\) and \(O\) atoms?
b. What electron configurations of the molecule are likely to be low in energy? Consider all reasonable orderings of the molecular orbitals. What are the states corresponding to these configurations?
c. What are the bond orders in each of these states?
d. The true ground state of \(BO\) is \(^2\Sigma\). Specify the +/- and u/g symmetries for this state.
e. Which of the excited states you derived above will radiate to the \(^2\Sigma\) ground state? Consider electric dipole radiation only.
f. Does ionization of the molecule to form a cation lead to a stronger, weaker, or equivalent bond strength?
g. Assuming that the energies of the molecular orbitals do not change upon ionization, what are the ground state, the first excited state, and the second excited state of the positive ion?
h. Considering only these states, predict the structure of the photoelectron spectrum you would obtain for ionization of \(BO\).
problem on vibration-rotation spectroscopy
37.
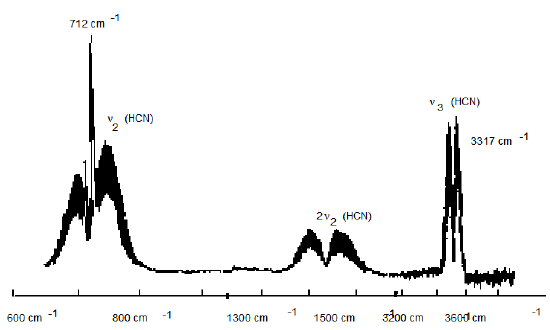
The above figure shows part of the infrared absorption spectrum of \(HCN\) gas. The molecule has a \(CH\) stretching vibration, a bending vibration, and a \(CN\) stretching vibration.
a. Are any of the vibrations of linear \(HCN\) degenerate?
b. To which vibration does the group of peaks between 600 cm-1 and 800 cm-1 belong?
c. To which vibration does the group of peaks between 3200 cm-1 and 3400 cm-1 belong?
d. What are the symmetries (s, p, d) of the \(CH\) stretch, \(CN\) stretch, and bending
vibrational motions?
e. Starting with \(HCN\) in its 0,0,0 vibrational level, which fundamental transitions would be infrared active under parallel polarized light (i.e., z-axis polarization):
\[000 \rightarrow 001?\]
\[000 \rightarrow 100?\]
\[000 \rightarrow010?\]
f. Why does the 712 cm-1 transition have a Q-branch, whereas that near 3317 cm-1 has only P- and R-branches?
Problem in Which You Can Practice Deriving Equations
This is Important Because a Theory Scientist Does Derivations as Part of Her/His Job
38.
By expanding the molecular orbitals {\(\phi_k\)} as linear combinations of atomic orbitals {\(\chi_\mu\)},
\[\phi_k =\sum_\mu c_{\mu k}\chi_\mu\]
show how the canonical Hartree-Fock (HF) equations:
\[F \phi_i = \varepsilon_i \phi_j\]
reduce to the matrix eigenvalue-type equation of the form:
\[\sum_\nu F_{\mu\nu}C_{\nu i}= \varepsilon_i\sum_\nu S_{\mu\nu}C_{\nu i}\]
where:
\[F_{\mu\nu} = \langle \chi_\mu|h|\chi_nu\rangle+\sum_{\delta\kappa} \left[\gamma_{\delta\kappa}\langle\chi_\mu\chi_\delta|g|\chi_\nu\chi_\kappa\rangle - \gamma_{\delta\kappa}^{\rm ex} \langle\chi_\mu\chi_\delta|g|\chi_\kappa\chi_\nu\rangle\right] ,\]
\[S_{\mu\nu} = \langle\chi_\mu|\chi_nu\rangle,\; \gamma_{\delta\kappa} =\sum_{i={\rm occ}}C_{\delta i}C_{\kappa i} ,\]
and
\[\gamma_{\delta\kappa}^{\rm ex} =\sum_{i=\text{occ and same spin}} C_{\delta i}C_{\kappa i}.\]
Note that the sum over \(i\) in \(\gamma_{\delta\kappa}\) and \(\gamma_{\delta\kappa}^{\rm ex}\) is a sum over spin orbitals. In addition, show that this Fock matrix can be further reduced for the closed shell case to:
\[F_{\mu\nu} = \langle \chi_\mu|h|\chi_nu\rangle +\sum_{\delta\kappa} P_{\delta\kappa}\bigg[\langle\chi_\mu\chi_\delta|g|\chi_\nu\chi_\kappa\rangle - \dfrac{1}{2} \langle\chi_\mu\chi_\delta|g|\chi_\kappa\chi_\nu\rangle\bigg] ,\]
where the charge bond order matrix, \(P\), is defined to be:
\[P_{\delta\kappa} =\sum_{i={\rm occ}} 2C_{\delta i}C_{\kappa i},\]
where the sum over \(i\) here is a sum over orbitals not spin orbitals.
Another Derivation Practice Problem
39. Show that the HF total energy for a closed-shell system may be written in terms of integrals over the orthonormal HF orbitals as:
\[E({\rm SCF}) = 2\sum_k^{\rm occ}\langle\phi_k|h|\phi_k\rangle+\sum_{k,l}^{\rm occ} [2\langle kl|g|kl \rangle - \langle kl|g|lk\rangle] +\sum_{\mu>\nu} \dfrac{Z_\mu Z_\nu}{Z_{\mu\nu}}\]
.
More Derivation Problem
40. Show that the HF total energy may alternatively be expressed as:
\[E({\rm SCF}) =\sum_k^{\rm occ}(\varepsilon_k+\langle\phi_k|h|\phi_k\rangle)+ \sum_{\mu>\nu}\dfrac{Z_\mu Z_\nu}{R_{\mu\nu}},\]
where the \(\varepsilon_k\) refer to the HF orbital energies.
Molecular Hartree-Fock SCF Problem
41. This problem will be concerned with carrying out an SCF calculation for the \(HeH^+\) molecule in the \(1\Sigma_g^+(^1\sigma^2)\) ground state. The one- and two-electron integrals (in atomic units) needed to carry out this SCF calculation at \(R = 1.4\) a.u. using Slater type orbitals with orbital exponents of 1.6875 and 1.0 for the \(He\) and \(H\), respectively are:
| S11 = 1.0, | S22 = 1.0, | S12 = 0.5784 |
| h11 = -2.6442, | h22 = -1.7201, | h12 = -1.5113, |
| g1111 = 1.0547, | g1121 = 0.4744, | g1212 = 0.5664, |
| g2211 = 0.2469, | g2221 = 0.3504, | g2222 = 0.6250, |
where 1 refers to \(1s_{He}\) and 2 to \(1s_H\). The two-electron integrals are given in Dirac notation. Parts a. – d should be done by hand. Any subsequent parts can make use of the QMIC software that can be found at
www.emsl.pnl.gov:2080/people/...a_nichols.html.
- Using \(\phi_1 \approx 1s_{He}\) for the initial guess of the occupied molecular orbital, form a 2x2 Fock matrix. Use the equation derived above in problem 38 for \(F_{\mu\nu}\).
- Solve the Fock matrix eigenvalue equations given above to obtain the orbital energies and an improved occupied molecular orbital. In so doing, note that \(\langle\phi_1|\phi_1\rangle= 1 = C_1^TSC_1\) gives the needed normalization condition for the expansion coefficients of the \(\phi_1\) in the atomic orbital basis.
- Determine the total SCF energy using the expression of problem 39 at this step of the iterative procedure. When will this energy agree with that obtained by using the alternative expression for \(E({\rm SCF})\) given in problem 40?
- Obtain the new molecular orbital, \(\phi_1\), from the solution of the matrix eigenvalue problem (part b).
- A new Fock matrix and related total energy can be obtained with this improved choice of molecular orbital, \(\phi_1\). This process can be continued until a convergence criterion has been satisfied. Typical convergence criteria include: no significant change in the molecular orbitals or the total energy (or both) from one iteration to the next. Perform this iterative procedure for the \(HeH^+\) system until the difference in total energy between two successive iterations is less than 10-5 a.u.
- Show, by comparing the difference between the SCF total energy at one iteration and the converged SCF total energy, that the convergence of the above SCF approach is primarily linear (or first order).
- Is the SCF total energy calculated at each iteration of the above SCF procedure as in problem 39 an upper bound to the exact ground-state total energy?
- Does this SCF wave function give rise (at \(R\rightarrow\infty\)) to proper dissociation products?
Configuration Interaction Problem
42. This problem will continue to address the same \(HeH^+\) molecular system as above, extending the analysis to include correlation effects. We will use the one- and two-electron integrals (same geometry) in the converged (to 10-5 au) SCF molecular orbital basis which we would have obtained after 7 iterations above. The converged MOs you should have obtained in problem 1 are:
\[\phi_1 = \left[\begin{array}{c}-0.89997792\\-0.15843012\end{array}\right] \phi_2 =\left[\begin{array}{c}-0.83233180\\1.21558030\end{array}\right] \]
a. Carry out a two configuration CI calculation using the \(1\sigma^2\) and \(2\sigma^2\) configurations first by obtaining an expression for the CI matrix elements \(H_{I,J}\) (\(I,J = 1\sigma^2, 2\sigma^2\)) in terms of one- and two-electron integrals, and secondly by showing that the resultant CI matrix is (ignoring the nuclear repulsion energy):
\[\left[\begin{array}{cc}-4.2720 & 0.1261 \\ 0.1261 & -2.0149\end{array}\right]\]
b. Obtain the two CI energies and eigenvectors for the matrix found in part a.
c. Show that the lowest energy CI wave function is equivalent to the following two-determinant (single configuration) wave function:
\[\dfrac{1}{2}\left\{|(\sqrt{a}\phi_1 + \sqrt{b}\phi_2) \alpha(\sqrt{a}\phi_1 - \sqrt{b}\phi_2)\beta| + |(\sqrt{a}\phi_1 - \sqrt{b}\phi_2)\alpha (\sqrt{a}\phi_1 + \sqrt{b}\phi_2)\beta|\right\}\]
involving the polarized orbitals: \(\sqrt{a}\phi_1 ± \sqrt{b}\phi_2\), where \(a = 0.9984\) and \(b = 0.0556\).
d. Expand the CI list to 3 configurations by adding \(1\sigma 2\sigma\) to the original \(1\sigma^2\) and \(2\sigma^2\) configurations of part a above. First, express the proper singlet spin-coupled \(1\sigma 2\sigma\) configuration as a combination of Slater determinants and then compute all elements of this 3x3 matrix.
\(E\). Obtain all eigenenergies and corresponding normalized eigenvectors for this CI problem.
f. Determine the excitation energies and transition moments for \(HeH^+\) using the full CI result of part e above. The nonvanishing matrix elements of the dipole operator \(\textbf{r}(x,y,z)\) in the atomic basis are:
\[\langle1s_H|z|1s_{He}\rangle= 0.2854 \text{ and } \langle1s_H|z|1s_H\rangle= 1.4.\]
First determine the matrix elements of \(\textbf{r}\) in the SCF orbital basis then determine the excitation energies and transition moments from the ground state to the two excited singlet states of \(HeH^+\).
g. Now turning to perturbation theory, carry out a perturbation theory calculation of the first-order wave function \(|1\sigma^2\rangle^{(1)}\) for the case in which the zeroth-order wave function is taken to be the \(1\sigma^2\) Slater determinant. Show that the first-order wave function is given by:
\[|1\sigma^2\rangle^{(1)} = -0.0442| 2\sigma^2\rangle.\]
h. Why does the \(|1\sigma2\sigma\rangle\) configuration not enter into the first-order wave function?
i. Normalize the resultant wave function that contains zeroth- plus first-order parts and compare it to the wave function obtained in the two-configuration CI study of part b.
j. Show that the second-order RSPT correlation energy, \(E^{(2)}\), of \(HeH^+\) is -0.0056 a.u. How does this compare with the correlation energy obtained from the two-configuration CI study of part b?
Repeating the SCF Problem but With a Computer Program
43. Using either programs that are available to you or the QMIC programs that you can find at the web site
www.emsl.pnl.gov:2080/people/...ichols_ja.html
calculate the SCF energy of \(HeH^+\) using the same geometry as in problem 42 and the STO3G basis set provided in the QMIC basis set library. How does this energy compare to that found in problem 42? Run the calculation again with the 3-21G basis basis provided. How does this energy compare to the STO3G and the energy found using STOs in problem 42?
Series of SCF Calculations to Produce a Potential Energy Curve
44. Generate SCF potential energy surfaces for \(HeH^+\) and \(H_2\) using the QMIC software or your own programs. Use the 3-21G basis set and generate points for geometries of \(R = 1.0, 1.2, 1.4, 1.6, 1.8, 2.0, 2.5,\) and \(10.0 a_0\). Plot the energies vs. geometry for each system. Which system dissociates properly?
Configuration Interaction Potential Curves for Several States
45. Generate CI potential energy surfaces for the 4 states of \(H_2\) resulting from a calculation with 2 electrons occupying the lowest 2 SCF orbitals (\(1\sigma_g\) and \(1\sigma_u\)) in all possible ways. Use the same geometries and basis set as in problem 44. Plot the energies vs. geometry for each system. Properly label and characterize each of the states (e.g., repulsive, dissociate properly, etc.).
Problem on Partition Functions and Thermodynamic Properties
46. F atoms have \(1s^2\,2s^2\,2p^5\) \(^2P\) ground electronic states that are split by spin-orbit coupling into \(^2P_{3/2}\) and \(^2P_{1/2}\) states that differ by only 0.05 eV in energy.
a. Write the electronic partition function (take the energy of the \(^2P_{3/2}\) state to be zero and that of the \(^2P_{1/2}\) state to be 0.05eV and ignore all other states) for each F atom.
b. Using , derive an expression for the average electronic energy of \(N\) gaseous F atoms.
c. Using the fact that \(kT=0.03\) eV at \(T=300\) °K, make a (qualitative) graph of \(\bar{E}/N\) vs \(T\) for \(T\) ranging from 100°K to 3000°K.
Problem Using Transition State Theory
47. Suppose that we used transition state theory to study the reaction
\(NO(g) + Cl_2(g) \rightarrow NOCl(g) + Cl(g)\) assuming it to proceed through a bent transition state, and we obtained an expression for the rate coefficient
\[k_{\rm bent}=\dfrac{kT}{h}e^{-E^\ne/kT}\dfrac{\dfrac{q^\ne}{v}}{\dfrac{q_{NO}}{v} \dfrac{q_{Cl_2}}{v}}\]
a. Now, let us consider what differences would occur if the transition state structure were linear rather than bent. Assuming that the activation energy \(E^\ne\) and electronic state degeneracies are not altered, derive an expression for the ratio of the rate coefficients for the linear and bent transition state cases
\[\dfrac{k_{\rm linear}}{k_{\rm bent}}\]
b. Using the following order of magnitude estimates of translational, rotational, and vibrational partition functions per degree of freedom at 300°K
\[q_t \sim 10^8, q_r \sim 10^2, q_v \sim 1,\]
what ratio would you expect for \(k_{\rm linear}/k_{\rm bent}\)?
Problem With Slater Determinants
48. Show that the configuration (determinant) corresponding to the \(Li^+\) \(1s(\alpha)1s(\alpha)\) state vanishes.
Another Problem With Slater Determinants and Angular Momenta
49. Construct the 3 triplet and 1 singlet wave functions for the \(Li^+\) \(1s^12s^1\) configuration. Show that each state is a proper eigenfunction of S2 and Sz (use raising and lowering operators for S2)
Problem With Slater Determinants for a Linear Molecule
50. Construct determinant wave functions for each state of the \(1\sigma^22\sigma^23\sigma^21\pi^2\) configuration of \(NH\).
Problem With Slater Determinants for an Atom
51. Construct determinant wave functions for each state of the \(1s^12s^13s^1\) configuration of \(Li\).
Problem on Angular Momentum of an Atom
52. Determine all term symbols that arise from the \(1s^22s^22p^23d^1\) configuration of the excited \(N\) atom.
Practice With the Slater Condon Rules
53. Calculate the energy (using Slater Condon rules) associated with the \(2p\) valence electrons for the following states of the \(C\) atom.
i. \(^3P(M_L=1,M_S=1),\)
ii. \(^3P(M_L=0,M_S=0),\)
iii. \(^1S(M_L=0,M_S=0)\), and
iv. \(^1D(M_L=0,M_S=0)\).
More Practice With the Slater Condon Rules
54. Calculate the energy (using Slater Condon rules) associated with the \(\pi\) valence electrons for the following states of the \(NH\) molecule.
i.\(^1\Delta\) \((M_L=2, M_S=0),\)
ii. \(^1\Sigma\) \((M_L=0, M_S=0),\) and
iii. \(^3\Sigma\) \((M_L=0, M_S=0).\)
Practice With The Equations of Statistical Mechanics
55. Match each of the equations below with the proper phrase A-K
\[\renewcommand{\arraystretch}{2.5}\begin{array}{|c|c|}\hline
B_2=-2\pi \int_0^\infty r^2 \bigg(\exp\bigg(-\dfrac{u(r)}{kT}\bigg)-1\bigg) dr & \phantom{\Bigg|}\hspace{2cm}\\\hline
\bar{E^2}-(\bar{E})^2=kT^2\Big(\dfrac{\partial E}{\partial T}\Big)_{N,V} &\phantom{\Bigg|} \\\hline
\dfrac{2\pi mKT}{h^2} &\phantom{\Bigg|} \\\hline
Q=\exp\bigg(-\dfrac{N\phi}{2kT}\bigg)\bigg(\dfrac{\exp(-\theta/2T)}{1-\exp(-\theta/2T)}\bigg)^{3N}&\phantom{\Bigg|} \\\hline
g(\nu)=a\nu^2 & \phantom{\Bigg|}\\\hline
Q=\dfrac{M!}{N!(M-N)!}q^N &\phantom{\Bigg|} \\\hline
\Theta=\dfrac{q \exp\bigg(\dfrac{\mu_0}{kT}\bigg)p}{1+q \exp\bigg(\dfrac{\mu_0}{kT}\bigg)p}& \phantom{\Bigg|}\\\hline
p_A=p_A^0X_A &\phantom{\Bigg|} \\\hline
\dfrac{c\omega}{kT}=-4 & \phantom{\Bigg|}\\\hline
W=W_{AA}N_{AA}+W_{BB}N_{BB}+W_{AB}N_{AB}&\phantom{\Bigg|} \\\hline
N_{AB}\cong \dfrac{N_A c N_B}{N_A+N_B} &\phantom{\Bigg|} \\\hline\end{array}\]
- Raoult's law
- Debye solid
- Critical Point
- Ideal adsorption
- Langmuir isotherm
- Bragg-Williams
- Partition function for surface translation
- Concentrated solution
- Fluctuation
- Virial coefficient
- Einstein solid
Problem Dealing With the Second Virial Coefficient
56. The Van der Waals equation of state is
\[\left(p+\bigg(\dfrac{N}{V}\bigg)^2a\right)(V-Nb)=NkT\]
solve this equation for \(p\), and then obtain an expression for \(\dfrac{pV}{NkT}\). Finally, expand \(\dfrac{pV}{NkT}\) in powers of \(\bigg(\dfrac{N}{V}\bigg)\) and obtain an expression for the second virial coefficient of this Van der Waals gas in terms of \(b\), \(a\), and \(T\).
Problem to Make You Think About Carrying Out Monte-Carlo and Molecular Dynamics Simulations
57. Briefly answer each of the following:
For which of the following would you be wisest to use Monte-Carlo (MC) simulation and for which should you use molecular dynamics (MD)
a. Determining the rate of diffusion of \(CH_4\) in liquid \(Kr\).
b. Determining the equilibrium radial distribution of \(Kr\) atoms relative to the \(CH_4\) in the above example
c. Determining the mean square end-to-end distance for a floppy hydrocarbon chain in the liquid state
Suppose you are carrying out a Monte-Carlo simulation involving 1000 \(Ar\) atoms. Further suppose that the potentials are pair wise additive and that your computer requires approximately 50 floating point operations (FPO's) (e.g. multiply, add, divide, etc.) to compute the interaction potential between any pair of atoms
d. For each M-C trial move, how many FPO's are required? Assuming your computer has a speed of 100 MFlops (i.e., 100 million FPO's per sec), how long will it take you to carry out 1,000,000 M-C moves?
e. If the fluctuations observed in the calculation of question d are too large, and you wish to make a longer M-C calculation to reduce the statistical "noise", how long will your new calculation require if you wish to cut the noise in half?
f. How long would the calculation of question d require if you were to use 1,000,000 \(Ar\) atoms (with the same potential and the same computer)?
g. Assuming that the evaluation of the forces between pairs of \(Ar\) atoms (\(\partial V/\partial r\);) requires approximately the same number of FPO's (50) as for computing the pair potential, how long (in sec) would it take to carry out a molecular dynamics simulation involving 1000 \(Ar\) atoms using a time step (\(\Delta t\)) of 10-15 sec and persisting for a total time duration of one nanosecond (10-9 sec) using the 100 MFlop computer?
h. How long would a 10-6 MFlop (i.e., 1 FPO per sec) Ph.D. student take to do the calculation in part d?
Problem to Practice Using Partition Functions
58. In this problem, you will compute the pressure-unit equilibrium constant \(K_p\) for the equilibrium
\[2Na \rightleftharpoons Na_2\]
in the gas phase at a temperature of 1000 K. Your final answer should be expressed in units of atm-1. In doing so, you need to consider the electronic term symbols of \(Na\) and of \(Na_2\), and you will need to use the following data:
i. \(Na\) has no excited electronic states that you need to consider.
ii. \(\dfrac{\hbar^2}{8\pi^2Ik} = 0.221\) K for \(Na_2\)
iii. \(\dfrac{h\nu}{k} = 229\) K for \(Na_2\)
iv. \(1 \text{ atm} = 1.01 \times 10^6 \text{ dynes cm}^{-2}\)
v. The dissociation energy of \(Na_2\) from the \(v\) = 0 to dissociation is \(D_0 = 17.3 \text{ kcal mol}^{-1}\).
a. First, write the expressions for the \(Na\) and \(Na_2\) partition functions showing their translational, rotational, vibrational and electronic contributions.
b. Next, substitute the data and compute \(K_p\), and change units to atm-1.
Problem Using Transition State Theory
59. Looking back at the \(NO+Cl_2\) reaction treated using transition state theory in Problem 47, let us assume that this same reaction (via. the bent transition state) were to occur while to reagents \(NO\) and \(Cl_2\) were adsorbed to a surface in the following manner:
a. both \(NO\) and \(Cl_2\) lie flat against the surface with both of their atoms touching the surface.
b. both \(NO\) and \(Cl_2\) move freely along the surface (i.e., they can translate parallel to the surface).
c. both \(NO\) and \(Cl_2\) are tightly bound to the surface in a manner that causes their movements perpendicular to the surface to become high-frequency vibrations.
Given this information, and again assuming the following order of magnitude estimates of
partition functions
\[q_t \sim 10^8, q_r \sim 10^2, q_v \sim 1\]
calculate the ratio of the TS rate constants for this reaction occurring in the surface adsorbed state and in the gas phase. In doing so, you may assume that the activation energy and all properties of the transition state are identical in the gas and adsorbed state, except that the TS species is constrained to lie flat on the surface just as are \(NO\) and \(Cl_2\).
Contributors and Attributions
Jack Simons (Henry Eyring Scientist and Professor of Chemistry, U. Utah) Telluride Schools on Theoretical Chemistry
Integrated by Tomoyuki Hayashi (UC Davis)