II. Defining Characteristics of Radical Addition Reactions
- Page ID
- 24614
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)A. Reaction Mechanisms
Radical addition can take place by either chain or nonchain reaction. For each of these there are two variations on the basic reaction mechanism. For chain reactions (Schemes 1 and 2) each variation has a different type of chain-transfer step. For nonchain reactions both mechanisms involve electron transfer. They differ in that the transfer is either from (Scheme 3) or to (Scheme 4) an organometallic complex.
1. Chain Reactions
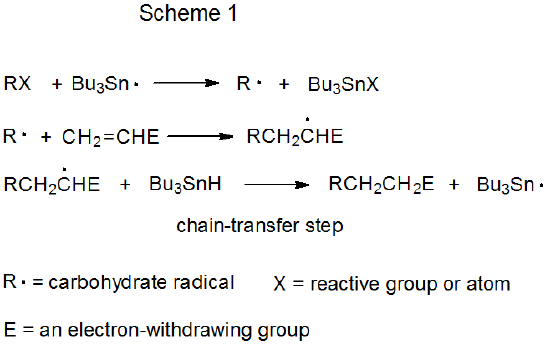
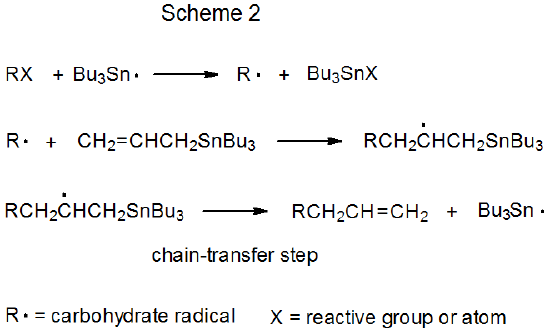
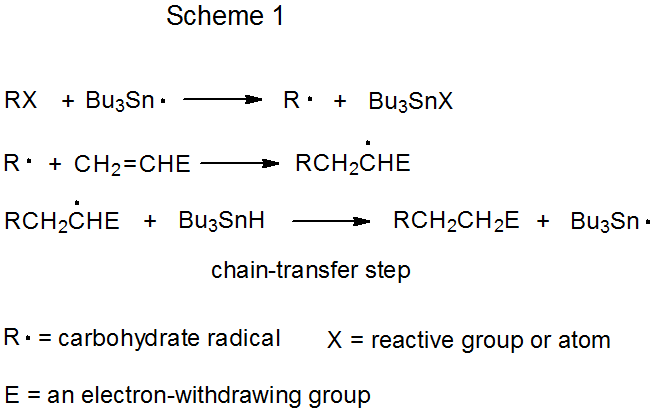
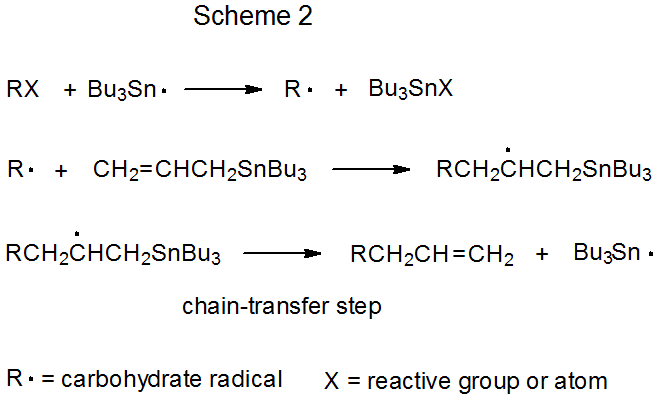
Both mechanisms for radical addition by chain reaction have a propagation phase that begins with group or atom abstraction (Schemes 1 and 2). In each of these mechanisms Bu3Sn· is shown as the abstracting radical, although other radicals [e.g., (Me3Si)3Si·] are capable of filling this role. The defining difference between these two mechanisms is the chain-transfer step. In the reaction shown in Scheme 1 it is bimolecular, and in that in Scheme 2 it is unimolecular.
{kind=link}
{kind=link}
a. Bimolecular Chain Transfer
The distinguishing feature of a chain reaction that takes place by bimolecular chain-transfer is an elementary reaction between a radical and a nonradical that ends one propagation sequence and creates the radical that begins a new sequence. In the reaction shown in Scheme 1, chain transfer occurs when the adduct radical abstracts a hydrogen atom from Bu3SnH to form the addition product RCH2CH2E and generate the chain-carrying radical Bu3Sn·.
b. Unimolecular Chain Transfer
Unimolecular chain transfer describes a reaction in which the chain-transfer step in a propagation sequence is an elementary reaction with a single reactant. The propagation steps for a typical, unimolecular, chain-transfer reaction are shown in Scheme 2, where the transfer step is a β-fragmentation that produces an unsaturated compound and a chain-carrying radical.
2. Nonchain Reactions
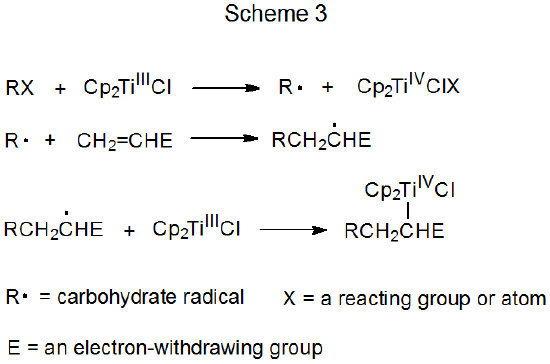
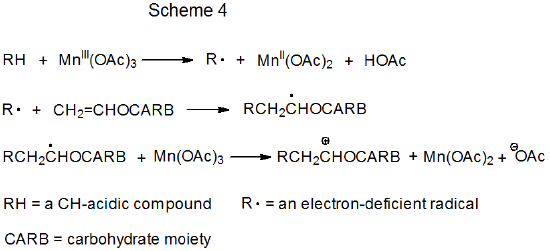
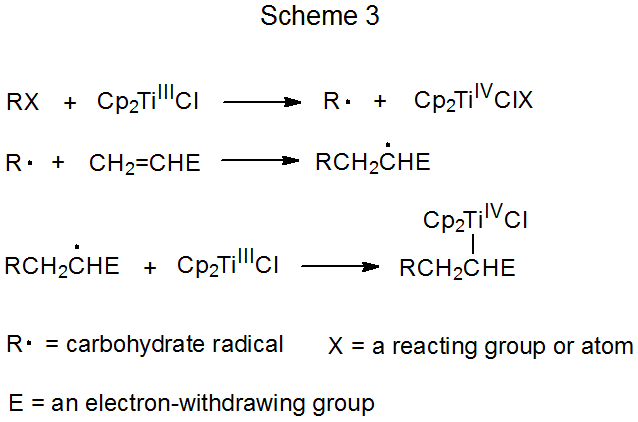
Schemes 3 and 4 each describe a mechanism for a reaction that has no repeating cycle. In the first of these (Scheme 3) the carbohydrate radical R· forms by electron transfer from a transition-metal complex (Cp2TiCl) to a carbohydrate derivative. (Cp2TiCl is one of several transition-metal complexes known to function as an electron donor in this type of reaction.) Radical reaction ends when a second molecule of Cp2TiCl combines with an adduct radical to produce an unstable, carbometallic product. Products of this type undergo rapid, nonradical reaction (e.g., elimination of the elements of Cp2TiClH to form a double bond).
{kind=link}
In the second type of nonchain reaction (Scheme 4) radical formation occurs when the transition-metal complex donates an electron to a CH-acidic compound. The radical phase of the reaction ends with a second electron transfer, one that produces a carbocation. This cation undergoes rapid, nonradical reaction, such as capture by a molecule of solvent.
{kind=link}
B. Selectivity in Addition Reactions
1. Chemoselectivity
Chemoselectivity is of consequence at two stages in a radical addition reaction. The first is in the radical forming step, where selectivity is determined by the reactivity of the functional groups present in a molecule of substrate. Forming the desired radical is accomplished at this stage by insuring that the radical precursor has the most reactive substituent attached to the carbon atom where the radical center is to be located. The next place at which selectivity potentially is of significance is during radical addition to the multiple bond. Chemoselectivity is meaningful at this time if there are two or more multiple bonds to which addition can occur.
2. Regioselectivity
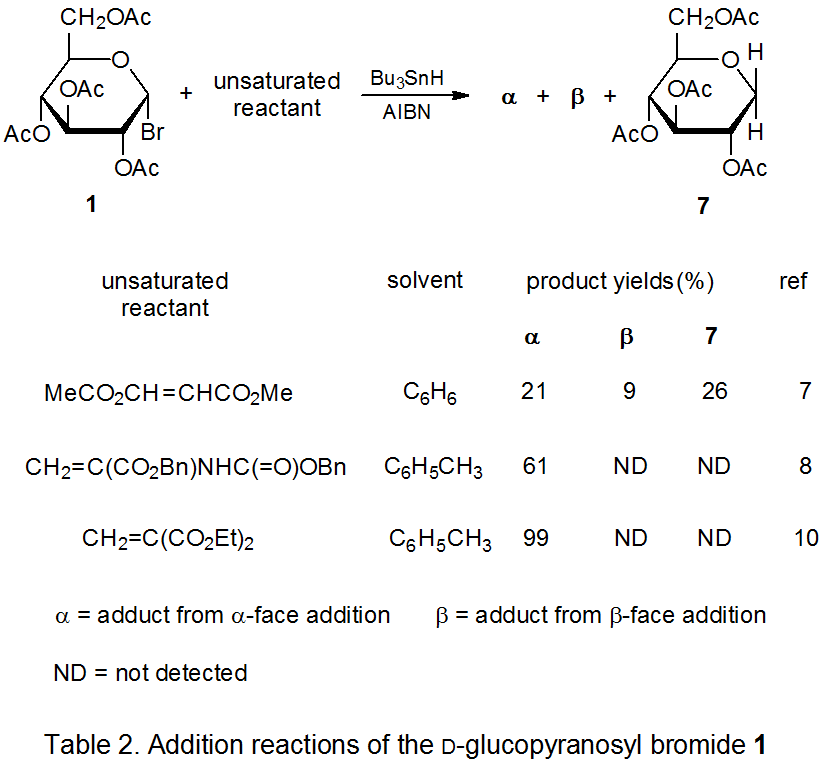
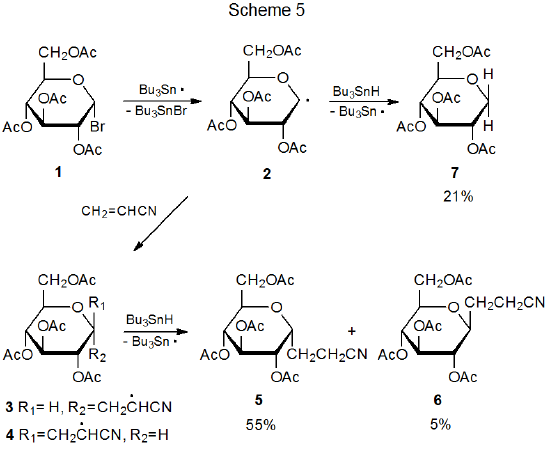
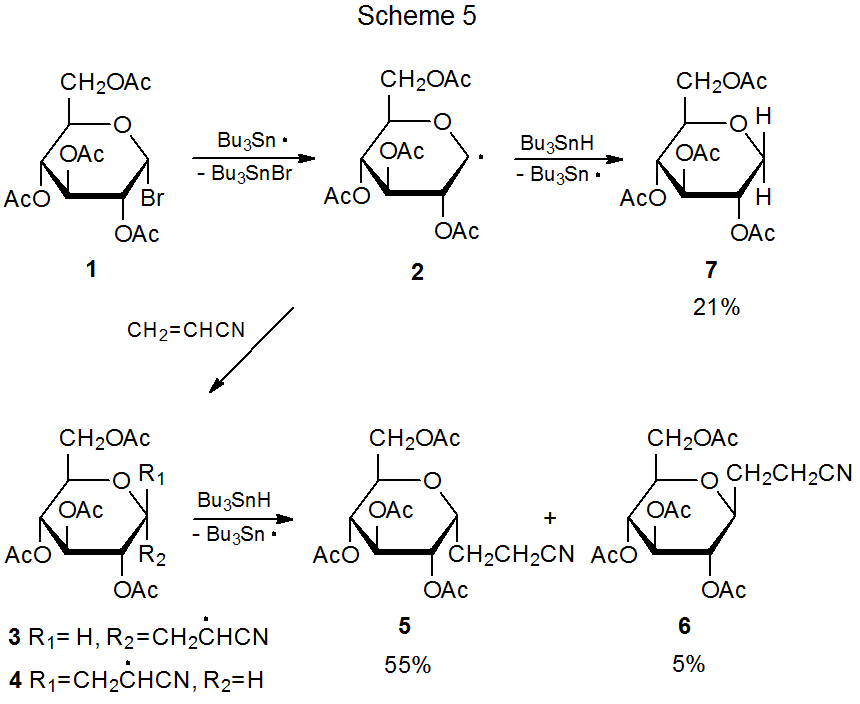
Most radical addition reactions involving carbohydrates are regiospecific. Addition occurs exclusively at the less substituted carbon atom in a multiple bond. The way in which the pyranos-1-yl radical 2 adds to acrylonitrile is typical (Scheme 5).1,2 The reactions in Tables 1 and 2 document a similar regiospecificity in the addition of 2 to other unsaturated compounds. The data in Tables 3-5 show that reactions of other pyranos-1-yl radicals exhibit similar reactivity. (All of these tables are located at the end of this chapter.)
{kind=link}
{kind=link}
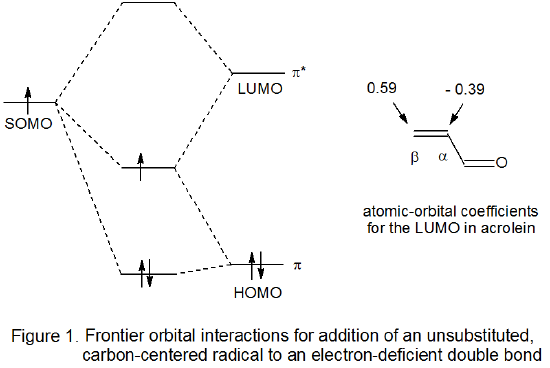
Both steric and polar effects have a role in determining regiospecificity in addition reactions. Steric effects become progressively more important as the effective size of the substituents near a multiple bond in an unsaturated compound increases and as the effective size of the adding radical increases. Polar effects exert themselves whenever a nucleophilic radical adds to an electron-deficient multiple bond, or an electrophilic radical adds to an electron-rich multiple bond. The extent of the influence of each effect depends upon the point at which the transition state is reached as a reaction progresses. For example, because addition of a nucleophilic radical to an electron-deficient multiple bond (the most common type of addition reaction) is exothermic, such a reaction should have an early (reactant-like) transition state11 with minimal, new bond formation. When there is little new bond formation at the transition state, there is a diminished opportunity for steric interactions to affect regioselectivity. The situation with polar effects is different because, as described in the next paragraph, they can exert considerable regioselective control in a reaction that has an early transition state.
For a reaction with an early transition state the molecular orbitals in the reactants do not change greatly by the time the transition state is reached. In such a situation frontier-orbital interactions are helpful not only in understanding why a reaction takes place but also in explaining reaction regioselectivity. Briefly, reaction occurs readily because the SOMO of the carbohydrate radical and the LUMO of the unsaturated reactant are energetically close enough for significant stabilizing interaction between the two at the transition state (Figure 1).12,13 The extent of early bonding between a radical and the carbon atoms in a reacting multiple bond is a function of the magnitude of the atomic orbital coefficient at each carbon atom in the LUMO of the unsaturated reactant. Attaching an electron-withdrawing or electron-donating substituent to a multiple bond causes these coefficients to be quite unequal, a situation that leads to regiospecific reaction. For a polarized multiple bond such as that found in an α,β-unsaturated nitrile or carbonyl compound, regiospecific addition to the β‑carbon atom reflects the greater magnitude of the atomic orbital coefficient at this atom in the LUMO when compared to the magnitude of the coefficient at the α carbon atom (Figure 1).
3. Stereoselectivity
As described in the next several sections, stereoselectivity in radical addition reactions is determined by a combination of steric and stereoelectronic effects. The stereoelectronic effect of primary importance is the kinetic anomeric effect. Steric effects that have an impact on reaction stereoselectivity have various names, but they all depend on steric interactions favoring a particular approach of an unsaturated compound or a hydrogen-atom transfer to a radical center.
a. The Kinetic Anomeric Effect
The reaction between the pyranos-1-yl radical 2 and an unsaturated compound with an electron-deficient double bond is highly stereoselective (Scheme 5). Preferential reaction on the α face of the pyranoid ring in 2 is due, in large part, to the kinetic anomeric effect (discussed in Chapter 11 of Volume I). Stereoselectivity in the addition reactions of 2 also is affected by reaction conditions. The information in Table 1 includes several sets of conditions suitable for highly stereoselective reaction with little, competing simple reduction.
{kind=link}
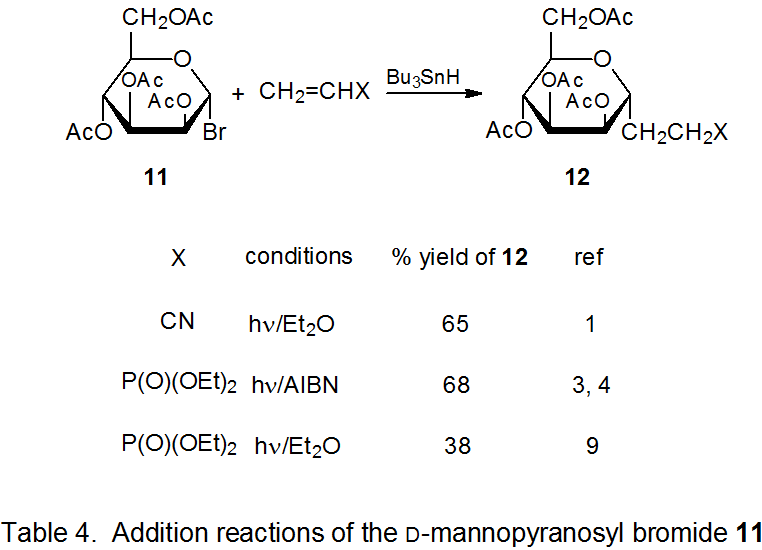
The selectivity observed in reactions of 2 extends to other D‑hexopyranos-1-yl radicals. Reactions of the D-galacto- and D-mannopyranosyl bromides 8 (eq 1) and 11 (eq 2), respectively, are at least as stereoselective as those of the corresponding D-glucopyranosyl bromide 1. The data in Tables 3 and 4 confirm that reactions of 8 and 11 occur preferentially on the α face of the pyranoid ring.
{kind=link}
{kind=link}
.png?revision=1&size=bestfit&width=430&height=133)
.png?revision=1&size=bestfit&width=370&height=127)
b. Steric Effects
(1). Group Shielding
Group shielding causes preferential addition to the less-hindered face of a ring system. In the reaction shown in eq 3 the 2,3-O-isopropylidene group shields the α face of the radical centered at C-4 in the pyranoid ring from approach by the unsaturated reactant.14
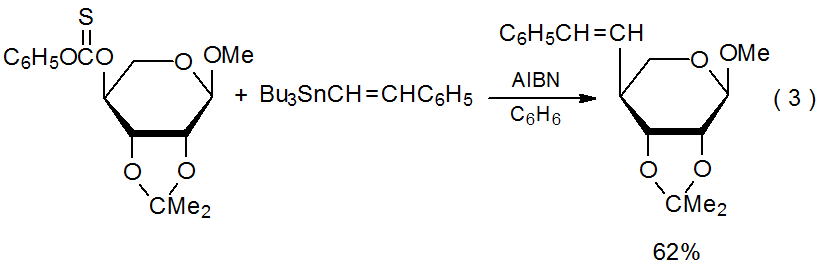.png?revision=1&size=bestfit&width=415&height=135)
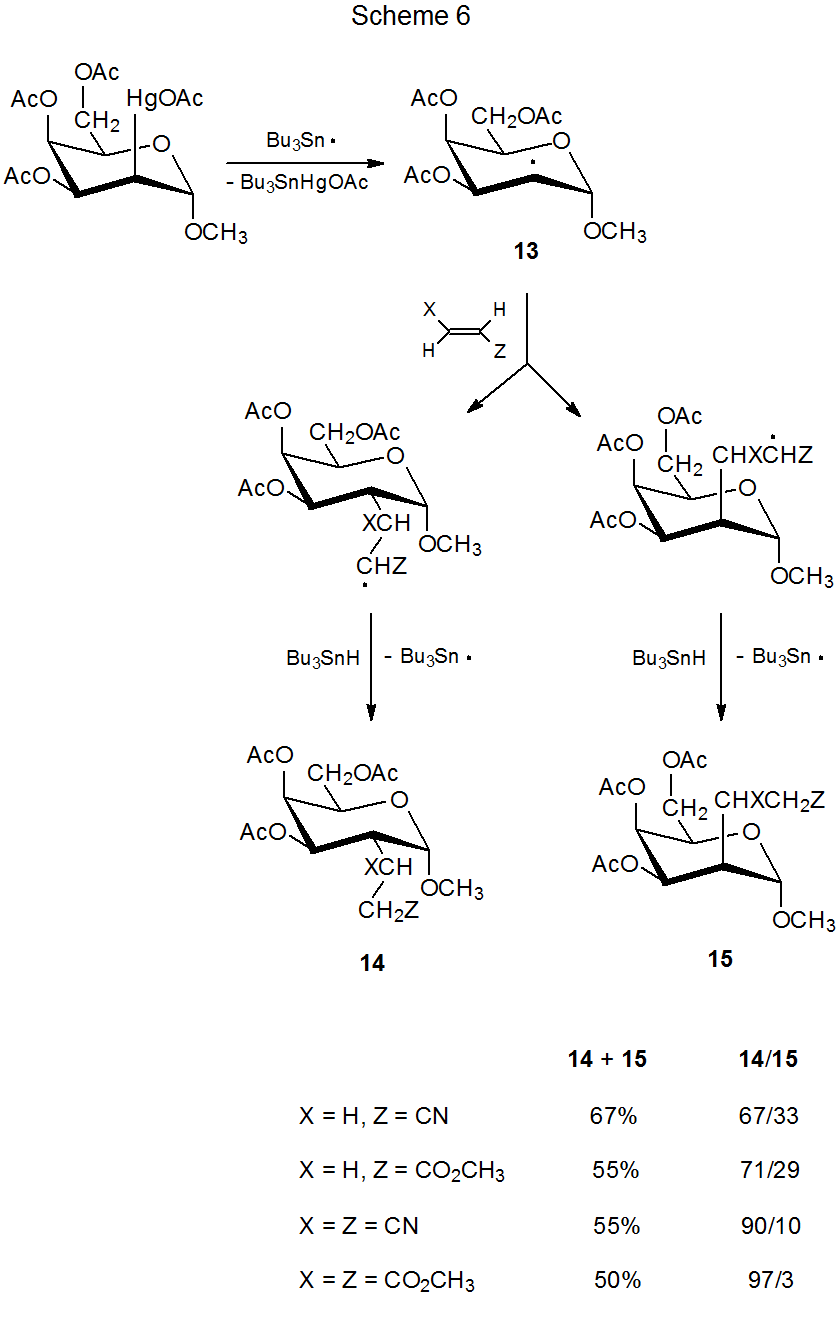
(2). Size of the Unsaturated Reactant
The reaction shown in Scheme 6 illustrates the combined effect of steric size of the unsaturated reactant and group shielding of a radical by ring substituents.15 In this reaction the amount of addition to the better shielded, β face of the pyranoid ring in the radical 13 decreases as the effective size of the unsaturated reactant increases. The data in Scheme 6 show that, even in a reaction with an early transition state, steric effects can have a significant role in determining reaction stereoselectivity if these effects are large enough.
(3). Size of the Hydrogen-atom transfer
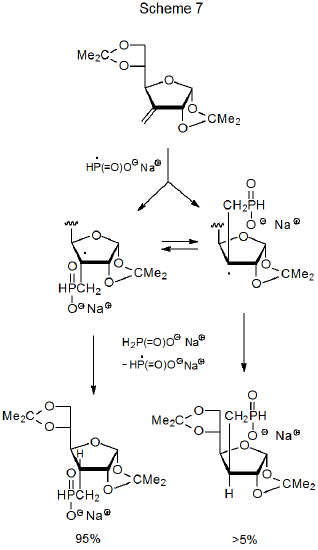
Sometimes a reaction forms the less stable of two possible stereoisomers due to restricted approach of a hydrogen-atom transfer to a radical center in the product-forming step. In the reaction shown in Scheme 7, for example, the less hindered approach of sodium hypophosphite to the β face of the furanoid ring produces the less stable stereoisomer in greater the 95% yield.16