III. Maximizing Transition-State Stabilization by Orbital Interactions: The Kinetic Anomeric Effect
- Page ID
- 23959
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)A. Radical Formation
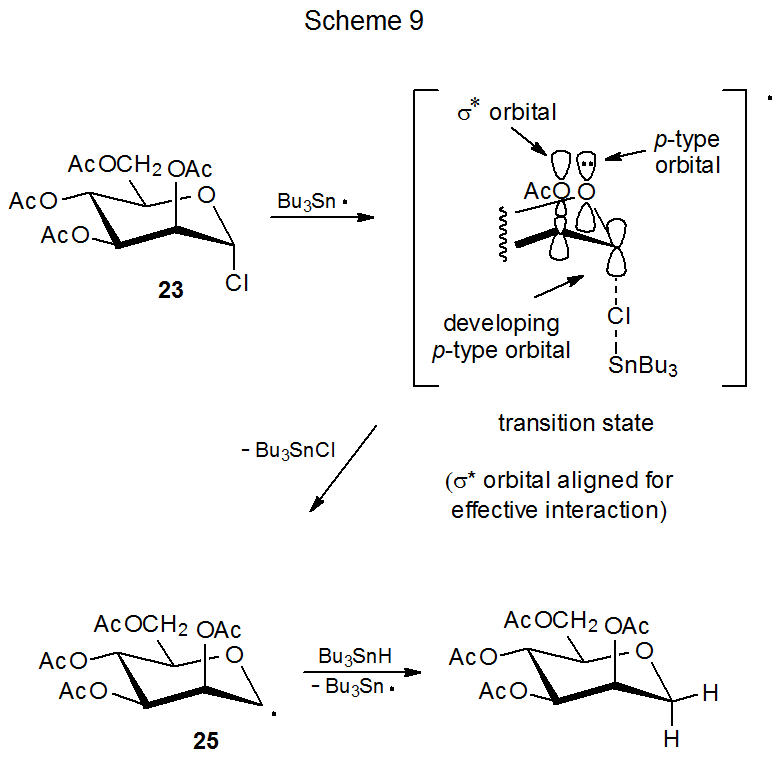
The reactions of the glycosyl chlorides 23 and 24 provide examples of stereoselectivity in pyranos-1-yl radical formation that depend on orbital interactions (Schemes 9 and 10).25,26 The difference in their reaction rates is linked to the orientation of the C2–O bond in each of these stereoisomers. The D-mannopyranosyl chloride 23 reacts with tri-n-butyltin hydride 7.8 times more rapidly than does the epimeric D-glucopyranosyl chloride 24. For 23 the C-2 acetoxy group has an orientation that allows transition-state stabilization by the orbital interactions shown in Scheme 9. These are the same interactions associated with the quasi-anomeric effect. (The quasi-anomeric effect, discussed in Section V of Chapter 6, provides an explanation for the conformations adopted by pyranos-1-yl radicals.27,28) Since these orbital interactions are developing at the transition state, they should be responsible, at least in part, for the greater reactivity of 23 when compared to 24. Such stabilization is minimal for reaction of 24 because the σ* orbital associated with the C2–O bond does not have the proper, transition-state orientation to assist significantly in stabilization (Scheme 10).
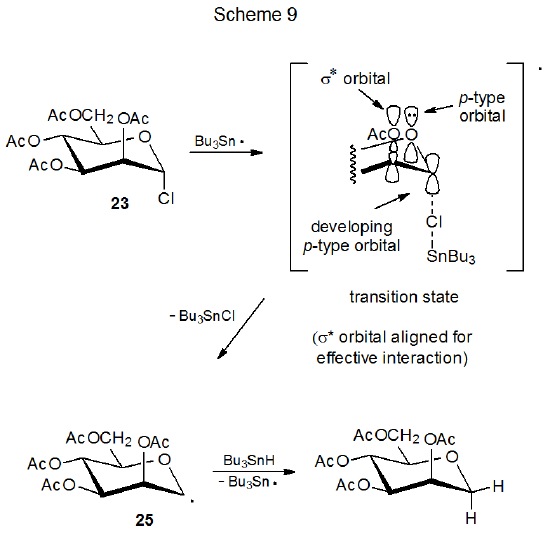
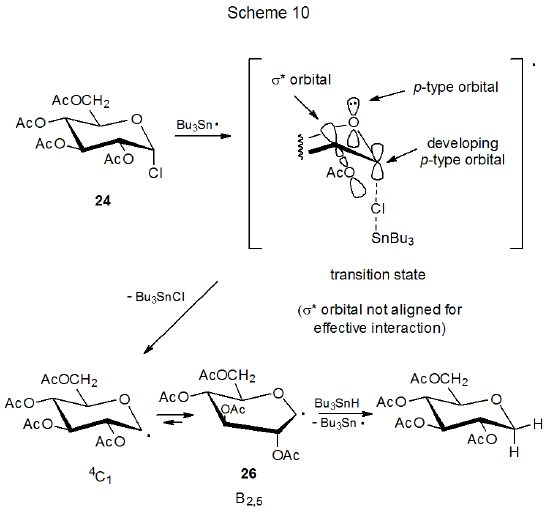
A critical assumption about the reactions shown in Schemes 9 and 10 is that the first step (chlorine-atom abstraction by the tri-n-butyltin radical), rather than the second step (hydrogen-atom abstraction from tri-n-butyltin hydride by the carbohydrate radical) is rate-determining. Such an assumption is reasonable because chlorine-atom abstraction in free-radical dehalogenation of alkyl chlorides is known to be rate-determining,29 but it is not a certainty because for reaction of iodides, bromides, and some very reactive chlorides, hydrogen-atom abstraction from tri-n-butyltin hydride is the rate-determining step.
B. Radical Reaction
1. The Role of Radical Conformation
a. The Curtin-Hammett Principle
Consider conformations X and Y of a radical that is undergoing an addition reaction to give the products Px and Py (Scheme 11). One possibility is that interconversion of X and Y is rapid compared to their reactions. In this situation the Curtin–Hammett principle applies; that is, the ratio of the products depends only on the relative energies of the transition states leading to their formation. (A more detailed statement of the Curtin-Hammet principle is “the relative amounts of products formed from two pertinent conformers are completely independent of the relative populations of the conformers and depend only on the difference in free energy of the transition states, provided the rates of reaction are slower than the rates of conformational interconversion”.30) An energy diagram showing a situation in which the Curtin-Hammet principle applies is found in Figure 2. If in this reaction Px and Py are stereoisomers, the overall process will form Py stereoselectively, even though conformer X is present in greater amount.
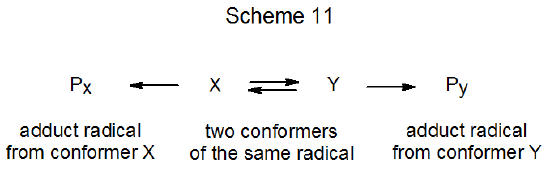
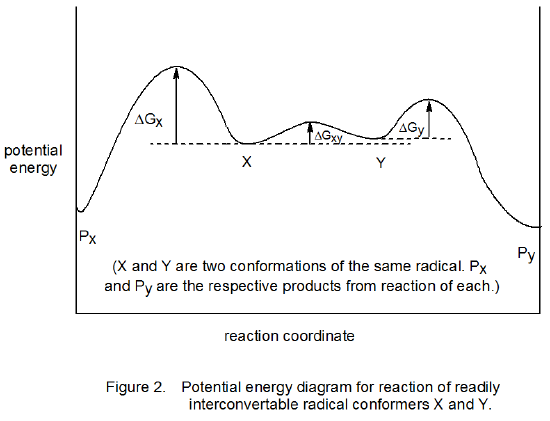
A different situation exists for the reaction shown in Figure 3. In this case the interconversion of radicals X and Y is slow compared to their reactions. Under these conditions (the Curtin–Hammett principle not in effect) the conformation present in greatest amount determines the stereoselectivity of the reaction; thus, if X is the major conformer, Px will be formed preferentially.
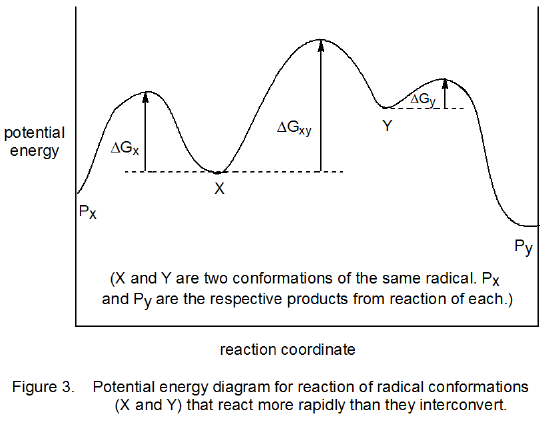
b. An Initial Proposal
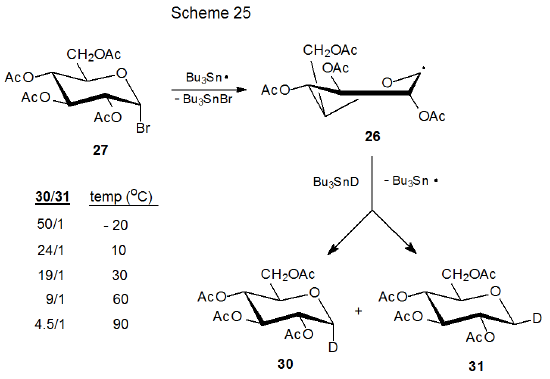
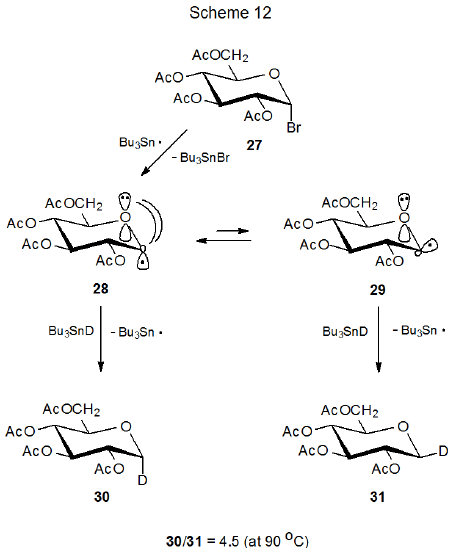
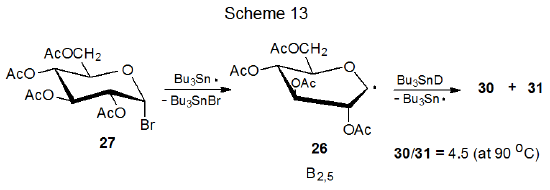
The explanation for the stereoselectivity of the reactions of pyranos-1-yl radicals has changed over time as new information about radical structure has become available. The first proposal for the stereoselective formation of the reduction products 30 and 31 from reaction of the D‑glucopyranosyl bromide 27 with tri-n-butyltin deuteride was that product yields reflected the relative amounts of 28 and 29 present in the reaction mixture (Scheme 12).31 (Inherent in this proposal was the assumption that 28 and 29 were the most stable structures for this pyranos-1-yl radical and that they reacted more rapidly than they equilibrated.) The major component in this proposed pseudoequilibrium was thought to be 28 because this radical would be stabilized more effectively than 29 by interaction of the orbital centered on C-1 with the p‑type orbital on the ring oxygen atom (Scheme 12). This interpretation subsequently had to be revised because ESR spectral analysis showed that neither 28 nor 29 was as stable as the distorted B2,5 boat conformation 26 (Scheme 13).27,28
c. A Revised Explanation
(1). Steric Effects
One possible explanation for stereoselectivity in reactions involving the D-glucopyranos-1-yl radical 26 is that steric effects are determining this selectivity just as they do in the reactions of many other carbohydrate radicals. For this explanation to be correct, approach to C-1 by Bu3SnD from the α face of 26 would have to be less hindered than a similar approach to its β face. With the neighboring 2-0-acetyl group shielding α-face reaction at C-1 (Scheme 13), it is difficult to see how the least hindered pathway to C-1 would involve approach to the α face of the radical.
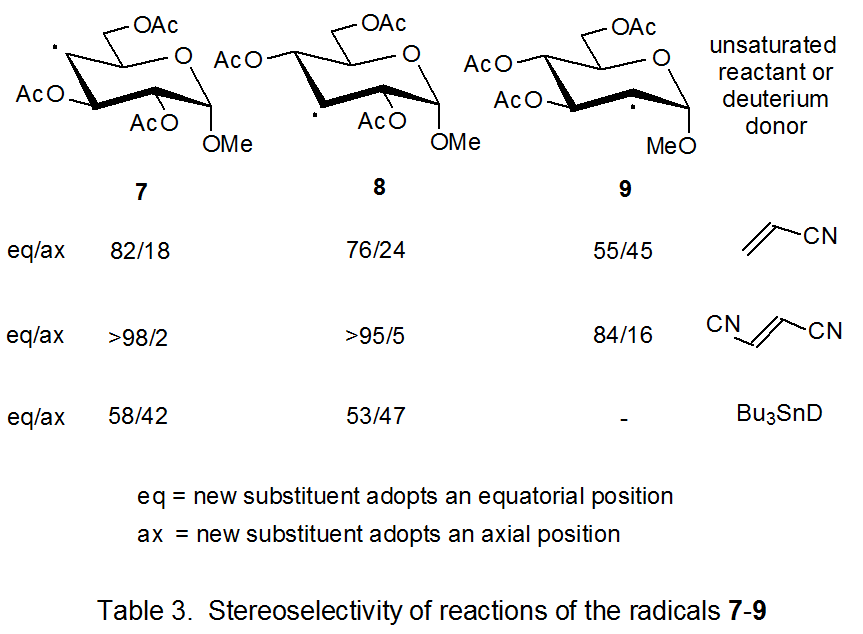
Another view of the difficulty with steric interactions being the controlling factor in stereoselectivity of the reactions of 26 comes from comparing these reactions with those of the pyranos-4-yl radical 7 and the pyranos-3-yl radical 8 (Table 3). For neither 7 nor 8 is the stereoselectivity of reaction with Bu3SnD large, but for each the major stereoisomer comes from reaction on the face of the radical opposite to that containing the shielding groups on the adjacent carbon atoms. This result stands in contrast to the reaction of the pyranos-1-yl radical 26, where the stereoselectivity is not only much larger, but Bu3SnD approaches this radical from the face containing the only shielding group on a neighboring carbon atom. The α‑face stereoselectivity of 26 is not limited to reaction with Bu3SnD. Addition of this radical to acrylonitrile also is stereoselectively from the more hindered α face (eq 4).
{kind=link}
.png?revision=1&size=bestfit&width=440&height=145)
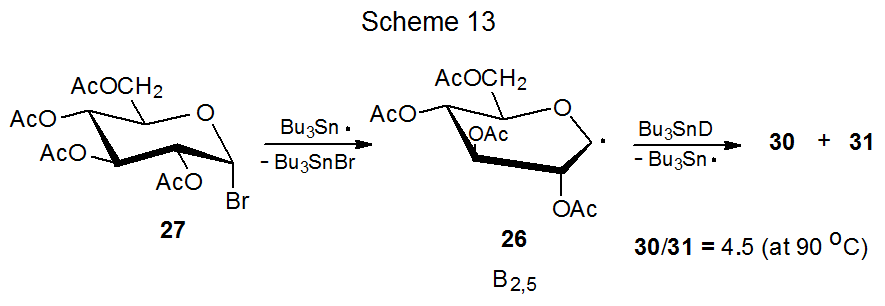
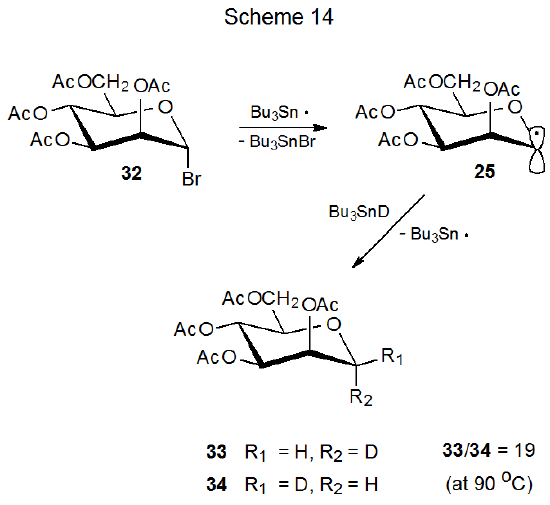
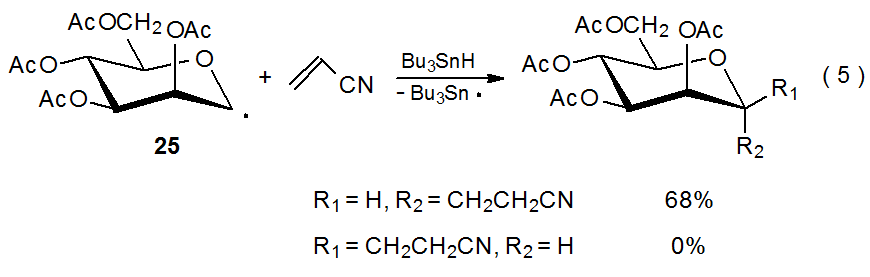
The D-mannopyranos-1-yl radical 25 exhibits even greater α-face selectivity in deuterium abstraction (Scheme 14) and addition to acrylonitrile (eq 5) than does its D-gluco epimer 26 (Scheme 13, eq 4). While steric effects could explain the stereoselectivity in the reactions of 25, they are unable to rationalize the selectivity in the reactions of both 25 and 26; however, for each of these radicals stereoselectivity does have a link to radical conformation.
{kind=link}
.png?revision=1&size=bestfit&width=435&height=139)
(2). Interconversion of Conformations
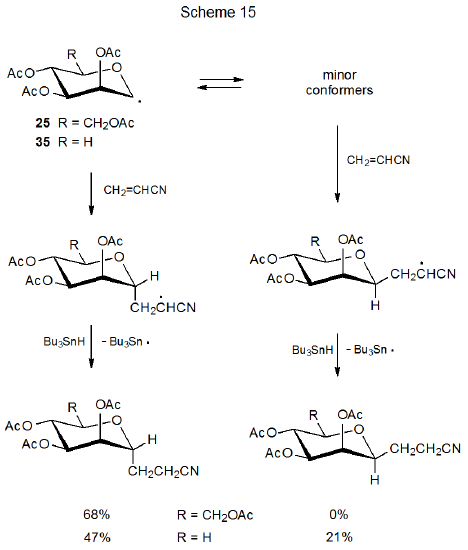
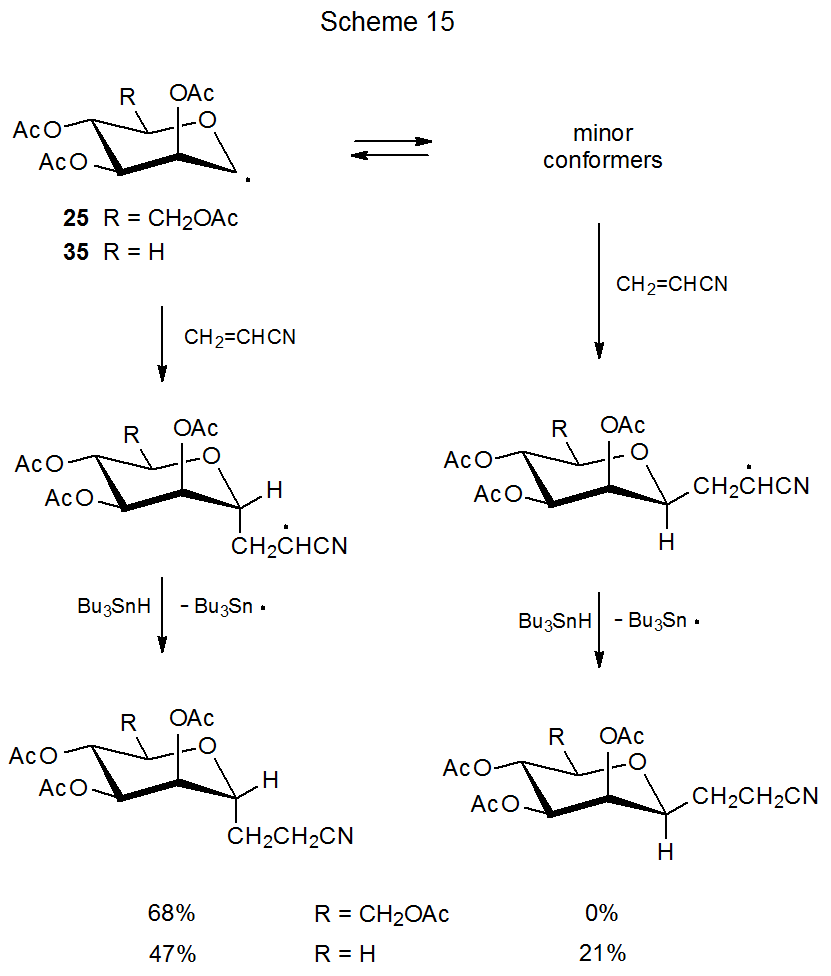
For the D‑mannopyranos-1-yl (25) and D‑lyxopyranos-1-yl (35) radicals only the 4C1 chair conformation can be detected in the ESR spectrum of each (Scheme 15).27,28,32 Structurally these radicals differ only at C-5 where the equatorial CH2OAc substituent in 25 is replaced by a hydrogen atom in 35. Even though the structures 25 and 35 are quite similar in the vicinity of the radical center, the stereoselectivity of their reactions is quite different. The radical 25 adds stereoselectively to acrylonitrile to give only the product arising from reaction at its α face, but the radical 35 reacts from both its α and β faces (Scheme 15).32
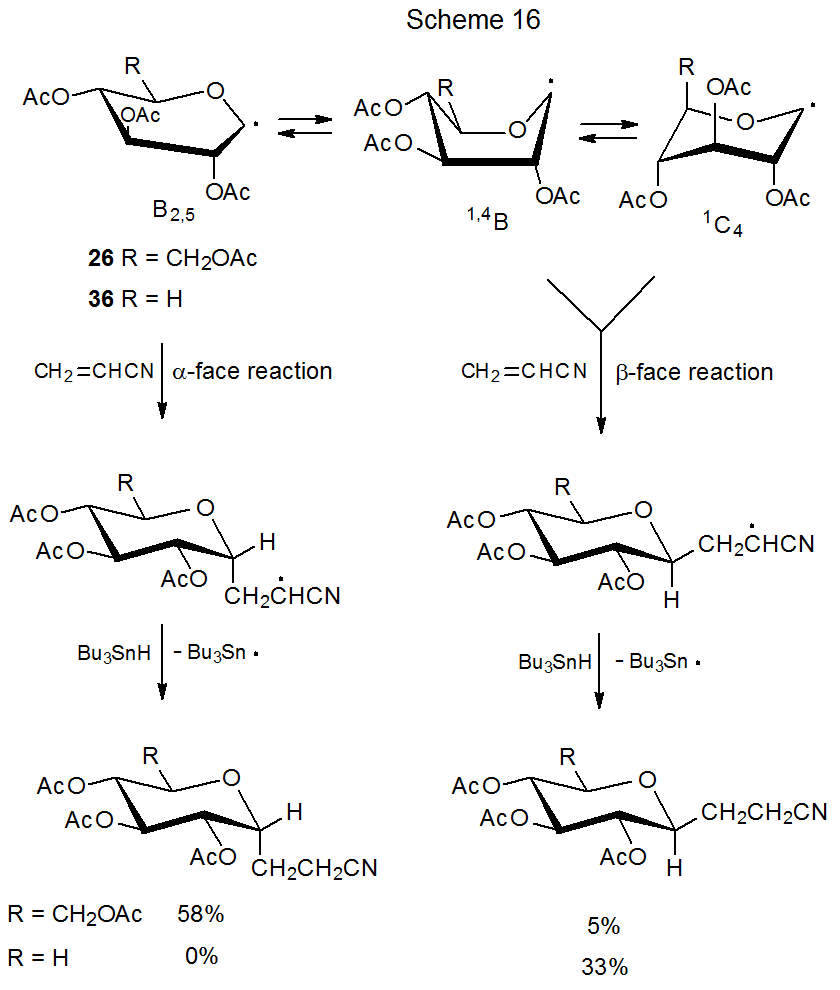
Since an equatorial substituent at C-5 is remote from the reacting center at C-1, steric effects alone cannot explain the difference in reactivity between these two (25 and 35). Findings concerning the reactivity of 25 and 35 (Scheme 15) are echoed in the reactions of the D-glucopyranos-1-yl radical 26 and the D‑xylopyranos-1-yl radical 36 (Scheme 16), further; observable conformations and reactivity 26 and 36 point to a possible explanation for the difference in stereoselectivity in the reactions of this pair (26 and 36) as well as the difference in stereoselectivity in the reactions of 25 and 35.
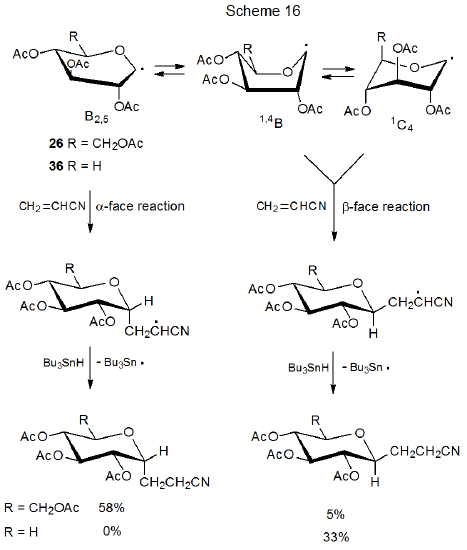
The ESR spectrum of 26 shows it to exist in a distorted B2,5 boat conformation, but for 36 B2,5 boat, 1,4B boat, and 1C4 chair conformations also can be detected.6 Conformational population, therefore, changes considerable when the CH2OAc substituent at C-5 in 26 is replaced by a hydrogen atom. This finding raises the possibility that the difference in stereoselectivity in the reactions of these radicals many be related to population and reactivity of conformational isomers (Scheme 16).
(3). Conformational Mobility
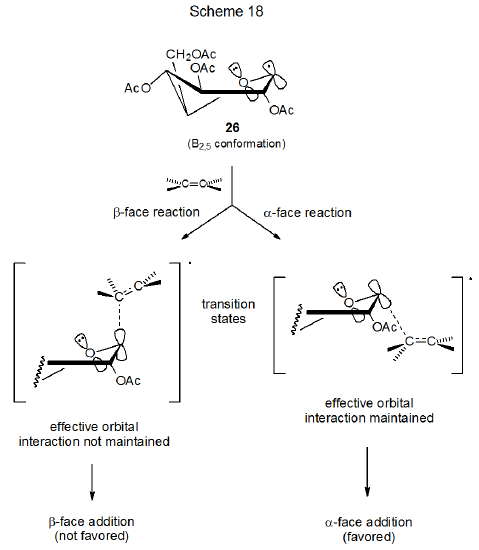
If B2,5 conformations react from their α face, and 1,4B or 4C1 conformers (or both) from their β face (Scheme 16), conformational population and mobility could explain the observed stereoselectivity in the reactions of radicals 26 and 36.6 According to this explanation, interconversion among conformers of the radical 36 would need to be more rapid than reactions of these radicals (Curtin-Hammett principle in effect). Since the assumption is that 1,4B boat or 1C4 conformers give β-face reaction products,6 the conclusion is that one or both of these conformers is much more reactive than the B2,5 conformation and that this very reactive conformation does so in a highly stereoselective fashion. A further part of this argument is that the CH2OAc substituent attached to C-5 in the radical 26 stabilizes the B2,5 conformation and, in so doing, increases its population to the point that little reaction occurs from other conformers (Scheme 16).
{kind=link}
A similar explanation exists for the difference in stereoselectivity between radicals 25 and 35 (Scheme 15). If interconversion among the conformers of the radical 35 is faster than reaction with acrylonitrile, it is possible to form a β-C-glycoside by reaction with a minor conformer. For the radical 25 the equatorial CH2OAc substituent at C-5 must stabilize the 4C1 conformation sufficiently that the concentration of minor conformers is too small for them to account for significant product formation.
{kind=link}
(4). Transition-State Stabilization: The Kinetic Anomeric Effect
To understand how radical conformation can have such a pronounced effect on stereoselectivity in the reactions of pyranos-1-yl radicals, it is instructive to examine transition-state stabilization. The major stereoisomer formed in these reactions results from a radical adopting a conformation that allows the stabilizing interaction between the p-type orbitals on C-1 and the ring oxygen atom to be maintained to the greatest extent in the transition-state. This conformation-dependent, stereoelectronic, transition-state stabilization is referred to as the kinetic33,34 or radical35,36 anomeric effect. (We will use “kinetic anomeric effect” in describing this phenomenon.)
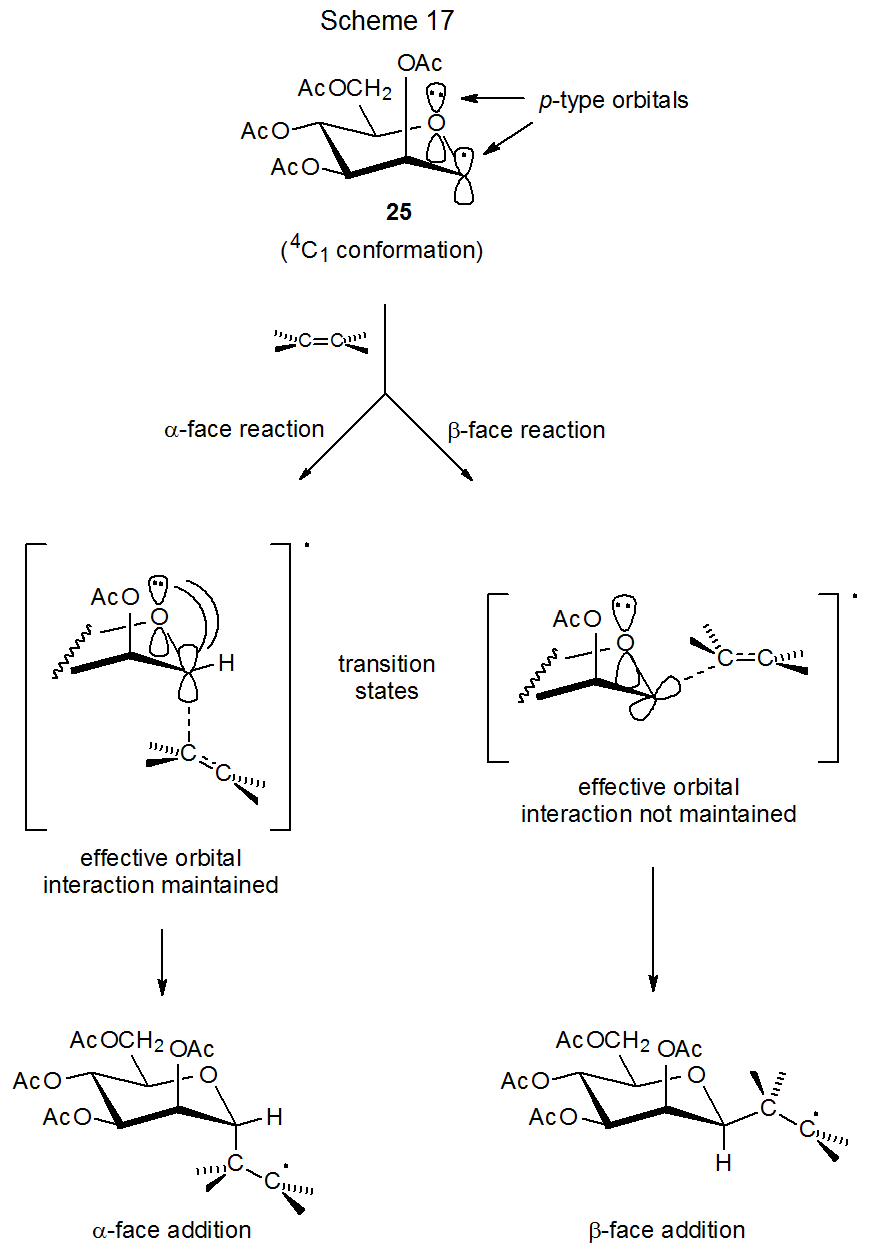
The kinetic anomeric effect offers a rationale for the D‑mannopyranos-1‑yl radical 25 adding to acrylonitrile exclusively from the α face of the pyranoid ring (Scheme 15). Only for α-face reaction will stabilizing interaction between the p-type orbitals on the ring oxygen atom and the singly occupied orbital on C-1 be maintained in the transition state (Scheme 17). Predicting stereoselectivity in the reaction of a conformationally mobile radical can be a challenging task because more than one conformation is accessible and determining which conformation is the most reactive may be difficult. This means that minor conformers, even ones that cannot be detected by ESR spectroscopy, can play a major role in determining reaction stereoselectivity. For the D-glucopyranos-1-yl radical 26 the only conformation detectable by ESR spectroscopy is a distorted B2,5 boat,27,28 but 1,4B and 1C4 conformations may be only modestly higher in energy and, therefore, easily accessible. For the B2,5 conformation, reaction from the α-face of the pyranoid ring maintains stabilizing orbital interaction more effectively than reaction from its β face (Scheme 18).
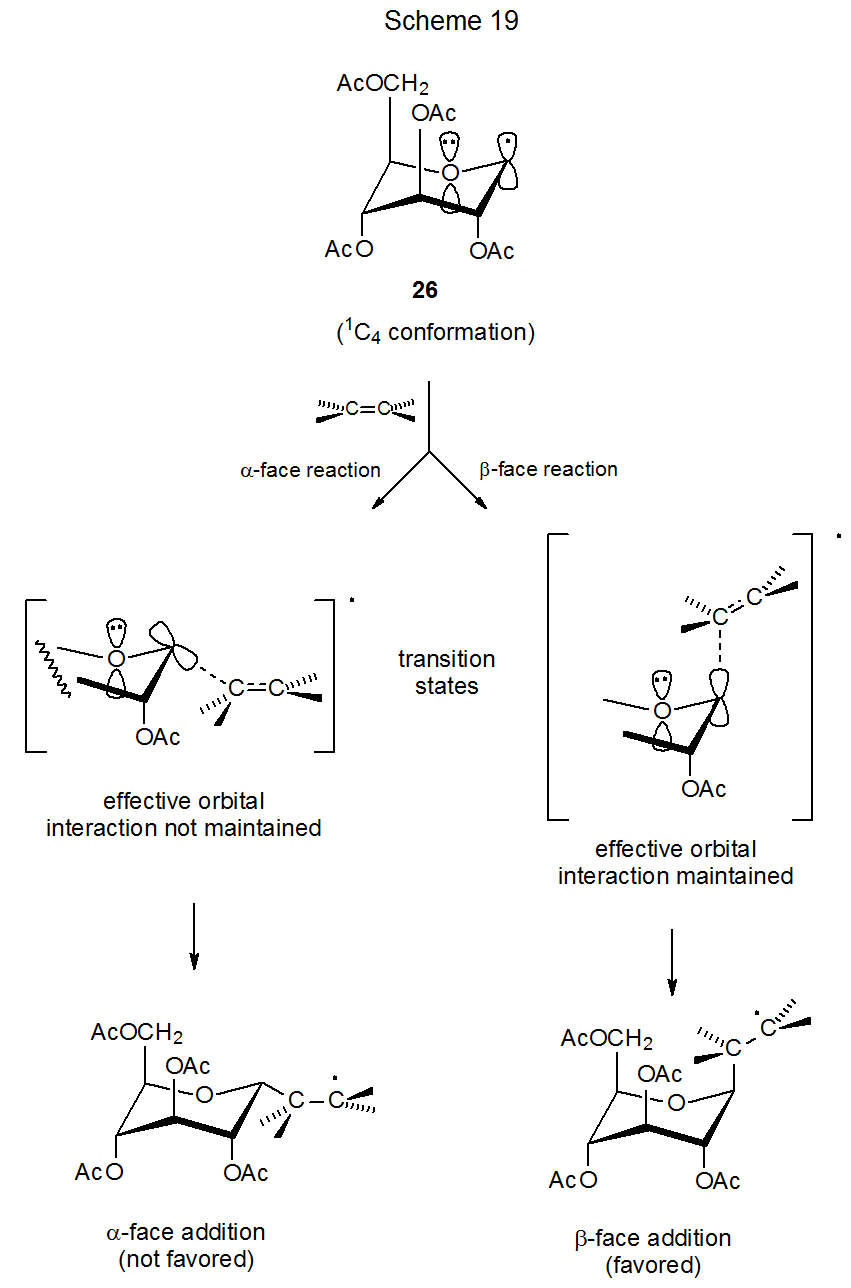
Reaction does occur, however, to a small extent (5%) from the β face of the pyranoid ring in the radical 26. The β anomer formed as a minor product could come from an accessible but undetected conformation, such as the 1C4. Maintaining orbital interactions in a 1C4 chair conformation would favor the β-face addition that leads to the minor product (Scheme 19). If this is the way in which β-face addition takes place, then stereoselectivity depends not only on which conformations are accessible and highly reactive but also on which face of a given conformer maintains the reaction-promoting orbital interaction that stabilizes the transition state.
Another explanation for the formation of the minor product in the reaction of the radical 26 is that this product results from a small amount of β‑face addition to the radical in its B2,5 conformation. Such an explanation, however, fails to explain greater β-face addition for the radical 36, when compared to 26 (Scheme 16), and is incompatible with the information, discussed in the next section, on reactions of radicals with restricted conformations.
Although the case is strong for radical conformation playing a critical role in stereoselectivity of reactions of pyranos-1-yl radicals, uncertainty remains about how to identify the reactive conformation when several may be present and undergoing reaction. This uncertainty is effecttively eliminated where radicals with restricted conformations are concerned. Study of such radicals has provided considerable insight into the power of the kinetic anomeric effect in determining reaction stereoselectivity.
(5). Restricted Conformations
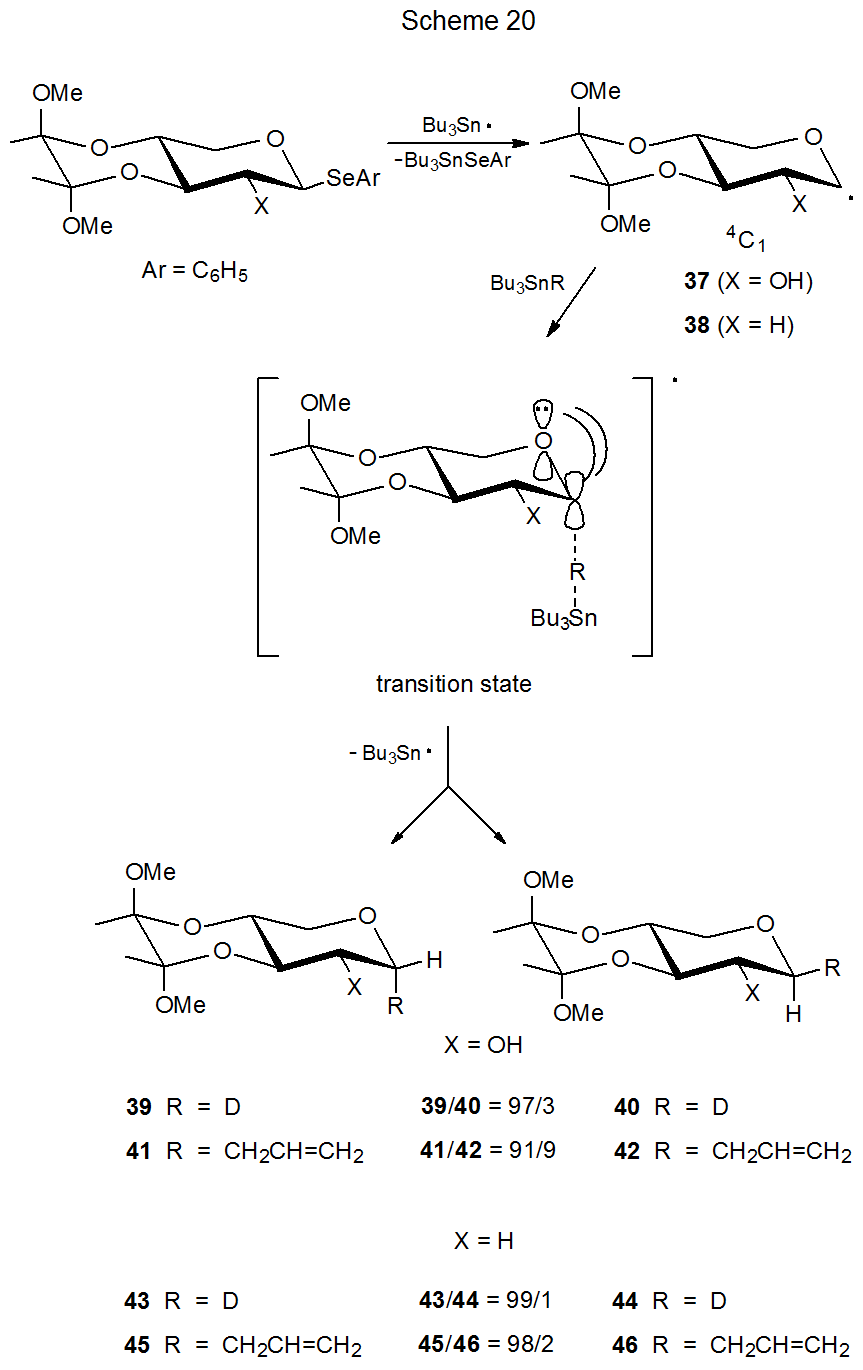
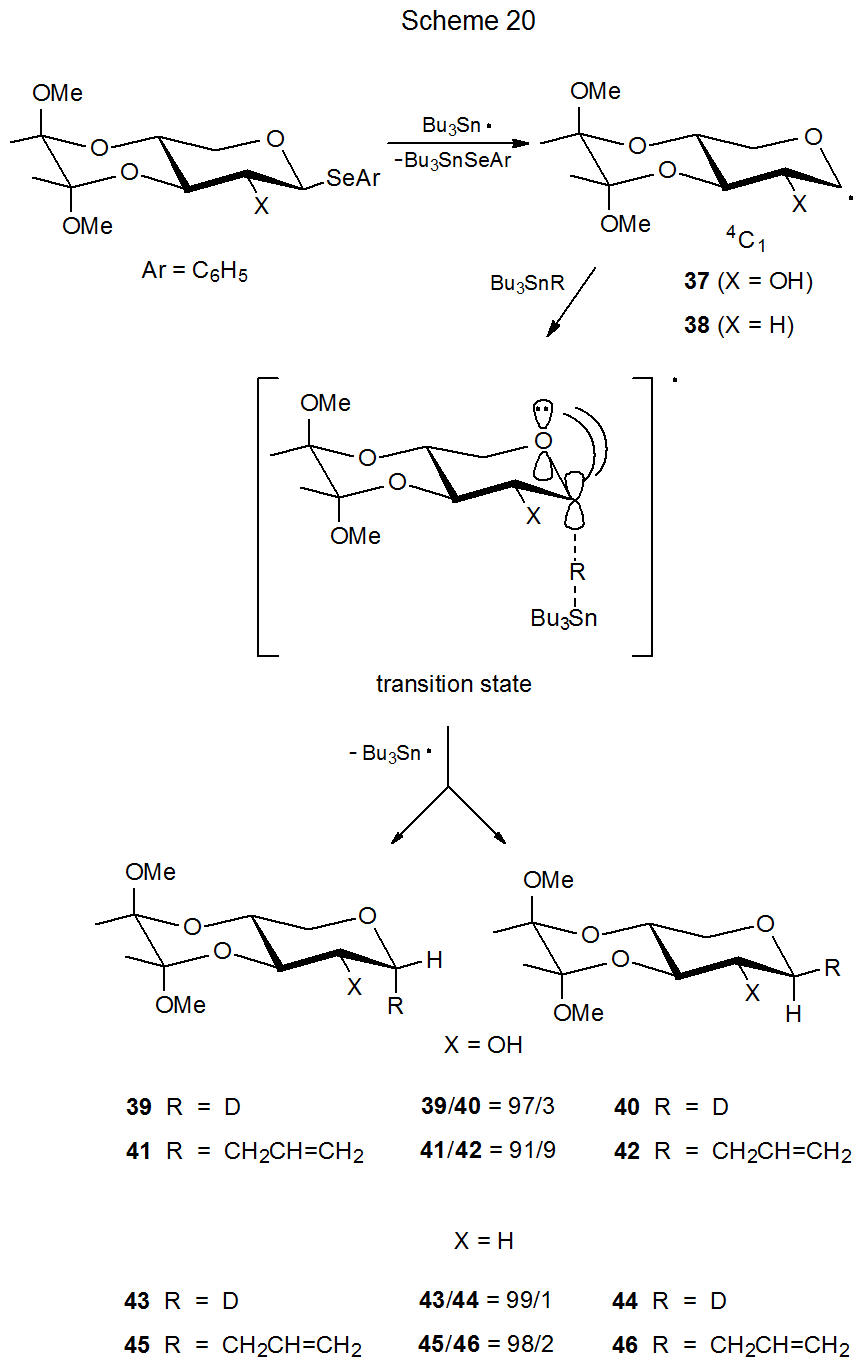
A restricted conformation for a radical is one that is highly favored over others due to some structural feature, such as an additional ring system. The radicals 37 (Scheme 20) and 47 (Scheme 21) have pyranoid rings restricted to 4C1 and 1C4 chair conformations, respectively. This restriction is caused by the presence of appropriately placed, additional rings.33 A second ring restricts conformational change in the radical 37 by creating a trans-decalin-type structure. For the radical 47 a bridge produces a bicyclic structure that creates a rigid, conformationally restricted system.
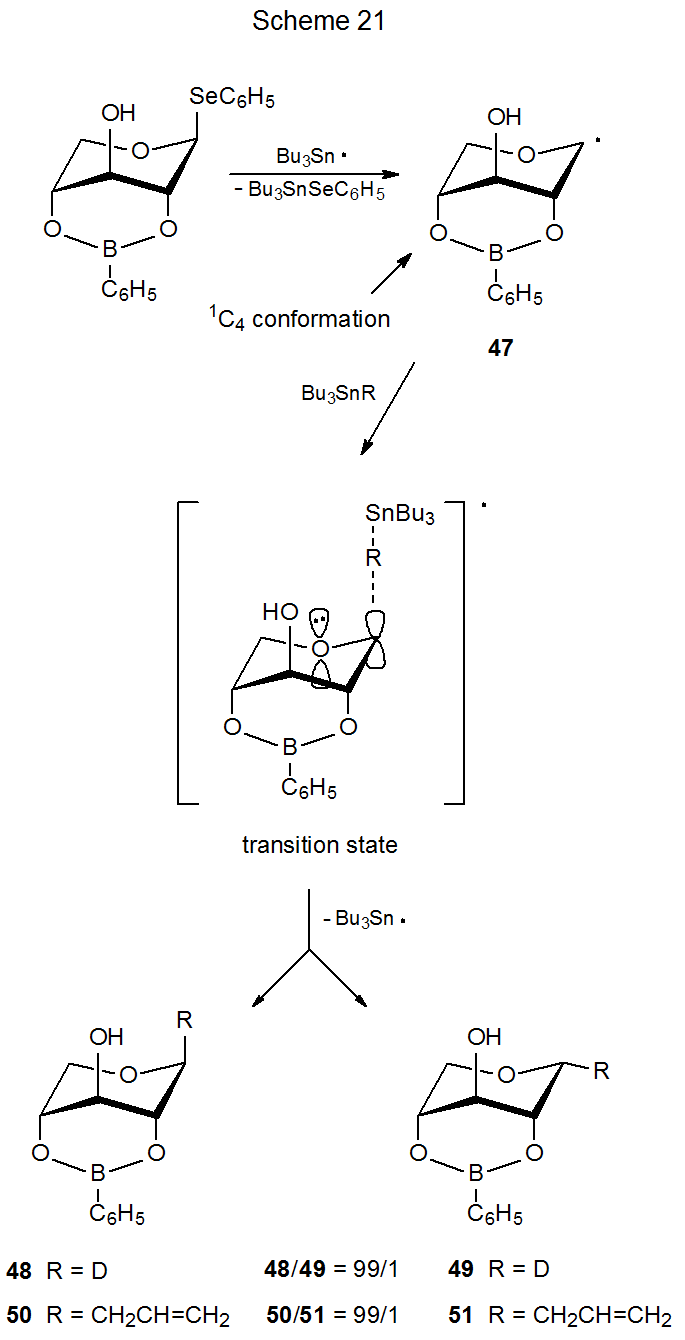
(a). Explanation for Reaction Stereoselectivity
For the radical 47 maintaining stabilizing orbital interaction in the transition state requires approach of a reacting molecule, such as tri-n-butyltin deuteride, to the β-face of the pyranoid ring (Scheme 21). This means that reaction proceeding according to the kinetic anomeric effect will give a product (48) with an axial deuterium atom at C-1. The high stereoselectivity of this reaction (48/49 = 99/1) supports the idea that conformational and stereoelectronic effects together have a powerful influence on the reactions of pyranos-1-yl radicals. Such an idea is reinforced in a dramatic fashion by the reaction of the radical 37 (Scheme 20). In this case in order to benefit from kinetic anomeric stabilization, the deuterium donor must approach the radical from the α face of the pyranoid ring. α‑Face selectivity (39/40 = 97/3) in this reaction (the pyranoid ring restricted to a 4C1 conformation) is nearly as great as the β‑face selectivity (48/49= 99/1) in the reaction of 47 (pyranoid ring restricted to a 1C4 conformation). Changing the radical conformation then completely changes reaction stereoselectivity;33 furthermore, the selectivity observed in each case is that predicted by the kinetic anomeric effect.
When Bu3SnCH2CH=CH2 replaces Bu3SnD as the molecule reacting with the radical 37, the stereoselectivity decreases somewhat, although it remains high.33 [The ratio of α-face to β-face reaction decreases from 97/3 (39/40) to 91/9 (41/42) (Scheme 20).] This modest decrease in stereoselectivity disappears when the hydroxy group at C-2 is replaced by a hydrogen atom: that is, the stereoselectivity for the reaction of the 2-deoxy radical 38 with Bu3SnD is essentially the same as that for reaction with Bu3SnCH2CH=CH2 (Scheme 20). Steric hindrance associated with the C-2 hydroxy group, therefore, may be responsible for the small difference in stereoselectivity observed for reactions of the radical 37 with Bu3SnD and Bu3SnCH2CH=CH2. The overall picture, however, remains one in which steric effects have, at most, a minor role in determining stereoselectivity in the reactions of these two radicals (37 and 38).
(b). σ*-Orbital Interaction
The σ* orbital of the C2–O bond in a pyranos-1-yl radical contributes to the stability of these radicals by interacting with the p-type orbitals on C-1 and the ring oxygen atom. Radical stabilization of this type plays a critical role in determining preferred radical conformation (see Section IV.A.2.d. of Chapter 6). A question raised by these orbital interactions concerns whether this type of stabilization also affects stereoselectivity in reactions of pyranos-1-yl radicals. The more rapid reaction of the D-mannopyranosyl chloride 23 (Scheme 9) when compared to the epimeric D‑glucopyranosyl chloride 24 (Scheme 10) indicates that it may. Even for 24, however, σ*-orbital interaction is not essential because reaction still takes place (although less rapidly) even with minimal participation from this orbital (Scheme 10). Study of radicals with restricted conformations helps to clarify the role of the C2–O σ* orbital in stereoselectivity of reactions of pyranos-1-yl radicals.
{kind=link}
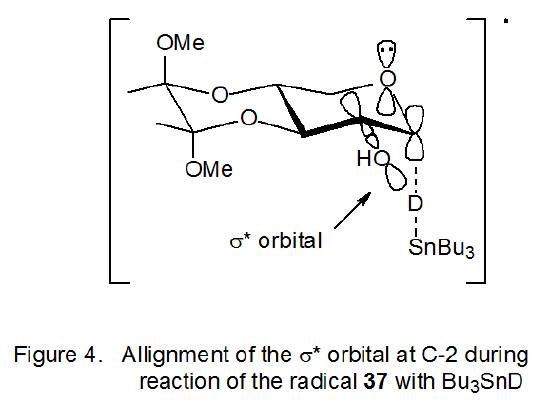
Because the pyranoid rings in radicals 37 and 38 (Scheme 20) are restricted to 4C1 conformations by their trans-decalin-like ring systems, the reactions of these radicals provide insight into the effect of a C2–O bond (in particular, its σ*-orbital) on the stereoselectivity of the reactions of pyranos-1-yl radicals. The radical 37, for example, reacts in a highly stereoselective fashion with tri-n-butyltin deuteride even though the σ* orbital of the C2–O bond is not aligned to assist in transition-state stabilization (Figure 4). The radical 38 completely lacks a C-2 substituent; yet, it also undergoes highly stereoselective reaction (Scheme 20). σ*-Orbital interaction involving the C2–O bond, therefore, is not essential to the stereoselectivity of the reactions of the radicals 37 and 38. The reactions of these radicals, however, point to the critical factor in determining reaction stereoselectivity, namely, maintaining effective interaction in the transition state between the singly occupied, p-type orbital on C-1 and the p-type orbital on the ring oxygen atom.
{kind=link}
Study of the reactions of radicals with restricted conformations generates a powerful argument in favor of the control that conformational and stereoelectronic effects have on the reactivity of pyranos-1-yl radicals. Among the radicals of this type discussed thus far, there is little indication that steric effects have other than a minor role in determining stereoselectivity. Study of compounds that have particularly well shielded radical centers, however, shows that steric effects can be significant in the stereoselectivity of some pyranos-1-yl radical reactions.
(6). Steric Effects Revisited
(a). Pyranos-1-yl Radicals
Even though conformational and stereoelectronic effects usually have the dominant role in determining stereoselectivity in the reactions of pyranos-1-yl radicals, comparing the reactions shown in equations 6 and 7 shows that steric effects can assert themselves when a sufficiently effective shielding group is present in a reacting molecule. Reaction of the glycosyl chloride 52 with allyltri-n-butyltin follows the now familiar pattern of α-face reaction that maintains transition-state interaction between orbitals on C-1 and the ring oxygen atom (eq 6).37 This reaction stands in contrast to that shown in eq 7 where the sterically demanding phthalimido group at C-2 causes a reversal of stereoselectivity in C-glycoside formation. This second reaction (eq 7) is a striking example of steric effects overwhelming the conformational and stereoelectronic effects that normally control stereoselectivity in reactions of pyranos-1-yl radicals.37,38
.png?revision=1&size=bestfit&width=435&height=132)
.png?revision=1&size=bestfit&width=440&height=167)
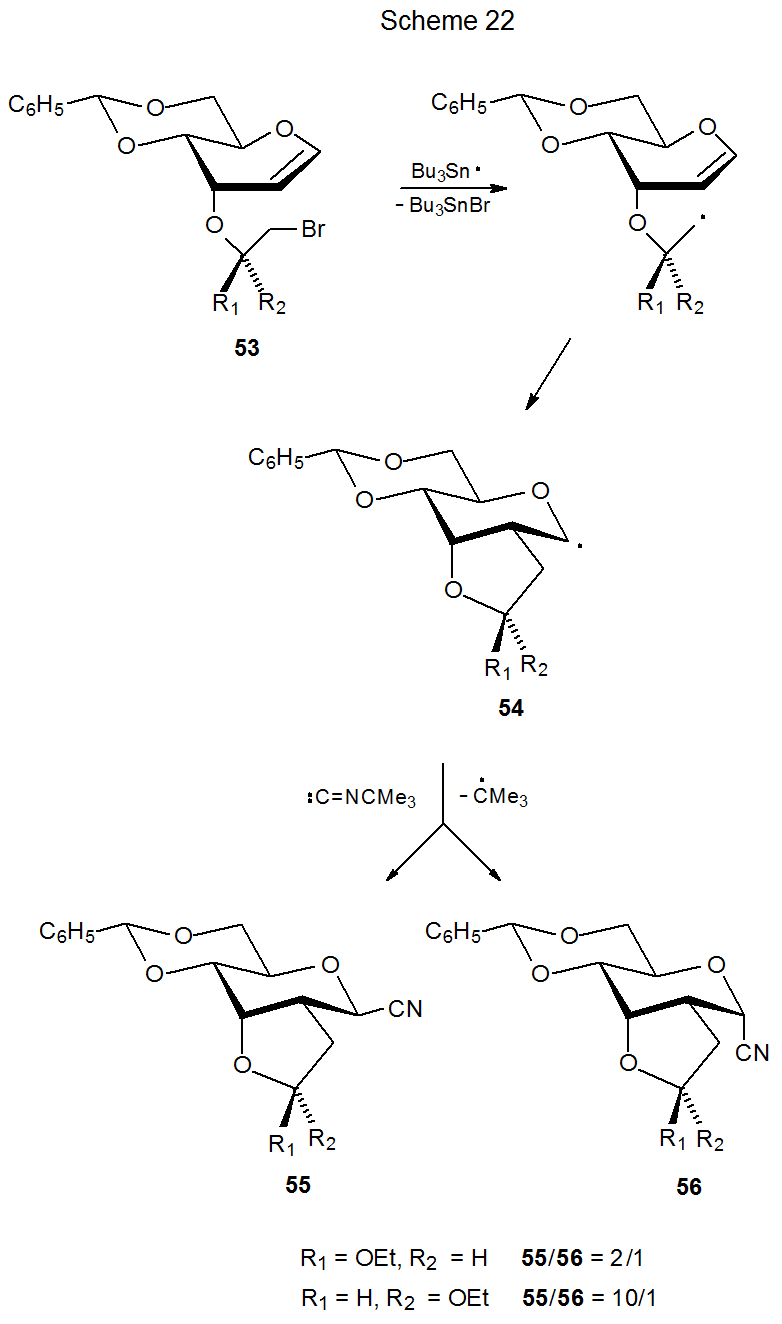
Reaction of the bromide 53 with tert-butyl isocyanide gives additional insight into the competition between steric and stereoelectronic effects in determining the stereoselectivity of reaction of pyranos-1-yl radicals (Scheme 22).35 The C-1 configuration in products 55 and 56 is determined by the stereoselectivity of the reaction between the radical 54 and tert-butyl isocyanide. In this reaction steric effects, which favor β-face reaction of 54, are more powerful than the stereoelectronic effects, which favor forming the product with an α configuration. In addition to shielding due to the methylene carbon atom attached to C-2, the shape of the radical in the vicinity of the ethoxy group has major impact on the steric effects directing the approach of tert-butyl isocyanide (Scheme 22).
(b). Furanos-1-yl Radicals
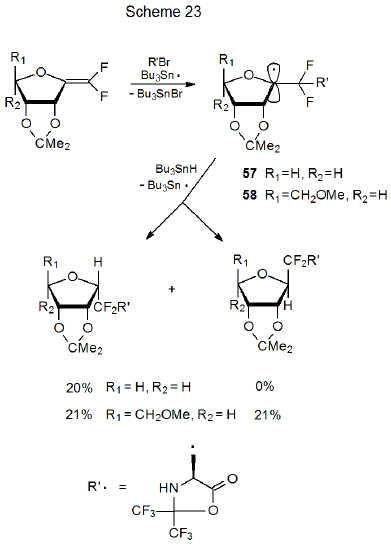
Study of pyranos-1‑yl radicals has identified conformational mobility as a “key” factor in the stereoselectivity of their reactions. This selectivity depends not only on which conformation is the major one but also on the accessibility and reactivity of other conformations. For furanos-1-yl radicals the same factors are involved, but their relative importance may differ. Conformational mobility is greater for furanos-1-yl radicals than for their pyranos-1-yl counterparts. Also, due to the shape of the furanoid ring, maintaining effective, transition-state interaction between p-type orbitals on C-1 and the ring oxygen atom (i.e., the interaction necessary for kinetic-anomeric stabilization) in many conformations is possible during reaction from both α and β faces of the ring. These observations lead to the proposal that conformational and stereoelectronic effects may be less important in determining stereoselectivity in furanos-1-yl radical reaction than in reactions of pyranos-1-yl radicals. The available information on stereoselectivity of the reactions of furanos-1-yl radicals is not sufficient to draw a firm conclusion about the relative importance of the kinetic anomeric effect when compared to steric effects. Exclusive hydrogen-atom transfer to the less-hindered β face of the radical 57 is consistent with steric effects controlling stereoselectivity; in addition, the fact that the radical 58, for which the β face is more hindered, is less stereoselective in its reaction also is consistent with steric effects primarily determining reaction stereoselectivity (Scheme 23).39
(c). Supersteric Radicals
Although bonding between two radicals normally is a rare event, such a reaction can become significant when other processes (e.g., hydrogen-atom abstraction or addition to an unsaturated compound) take place too slowly. Bonding between two radicals begins at a sufficiently long distance that differences in transition-state energies leading to different products sometimes are too small to be of consequence; as a result, little or no stereoselectivity is observed (eq 8).40
.png?revision=1&size=bestfit&width=420&height=147)
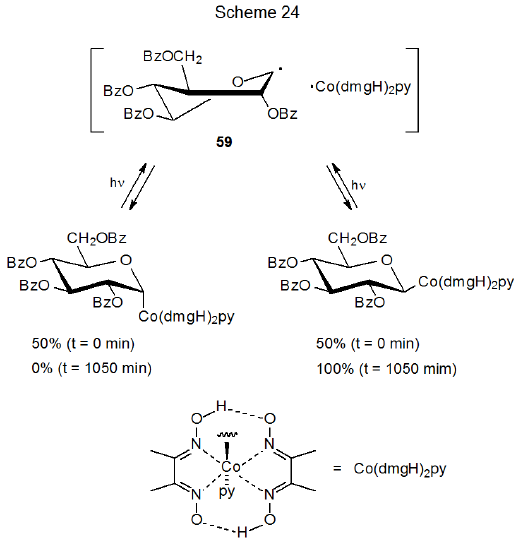
If one of the radicals involved in a coupling process has steric requirements that are great enough, even radical coupling becomes stereoselective. The "supersteric" Co(dmgH)2py radical, for example, has such a large steric requirement that its reaction with the pyranos-1-yl radical 59 occurs much more rapidly from the β face of the pyranoid ring in 59 than from its α face (Scheme 24).41 Having the Co(dmgH)2py substituent in an equatorial position (β anomer) is so much more stable than having it in the more crowded axial orientation (α anomer) that this difference significantly affects the transition-state energies leading to these two anomers. The steric size of the Co(dmgH)2py substituent is so great that it overcomes the usually more important kinetic anomeric effect in determining reaction stereoselectivity.
2. Effect of Temperature on Stereoselectivity
A final point with respect to stereoselectivity in bimolecular radical reactions concerns reaction temperature. In the process shown in Scheme 25 stereoselectivity is determined by the approach of Bu3SnD to the intermediate pyranos-1-yl radical 26.31 According to the kinetic anomeric effect [Section III.B.1.c.(4).] stereoselective deuterium transfer to the α face of 26 (to give 30) should have a lower transition-state energy than transfer to its β face (to give 31). At low temperature the stereoselectivity is high, but as the temperature rises, stereoselectivity decreases because a progressively larger percentage of radicals are able to react with Bu3SnD and transcend the barriers leading to both products (30 and 31). The results shown in Scheme 25 can be viewed as a general response of reaction stereoselectivity to changes in temperature.