
III. Radical Reactions
- Page ID
- 24004
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)A. Simple Reduction
1. Typical Reaction Conditions
Simple reduction of a halogenated carbohydrate is a reaction in which the only change that occurs in the substrate is replacement of a halogen atom with a hydrogen atom. This change typically is brought about by heating a solution of a halogenated carbohydrate, tri-n-butyltin hydride, and a catalytic amount of 2,2'-azobis(isobutyronitrile) (AIBN) in benzene or toluene at 80-110 oC for a few hours.
2. Thermodynamic Driving Force for Reaction
There is a powerful thermodynamic driving force for simple reduction of halogenated compounds with tri-n-butyltin hydride.14 This driving force is apparent in the highly exothermic reaction of bromomethane with tri-n-butyltin hydride (eq 6).14 The exothermic nature of this reaction derives from a carbon–bromine bond being replaced by a stronger carbon–hydrogen bond and a tin–hydrogen bond being exchanged for a stronger tin–bromine bond. The data in eq 7 show that simple reduction of chloromethane is also quite exothermic.14
.png?revision=1&size=bestfit&width=405&height=120)
.png?revision=1&size=bestfit&width=405&height=120)
3. Synthesis of Deoxygenated Sugars and Nucleosides
Simple reduction is a common reaction for halogenated sugars and nucleosides. A typical example, one in which halogen replacement takes place at C-4 in a pyranoid ring, is shown in eq 8.23 Other reactions, ones where the halogen atom is located at C-1,24,25 C‑2,26,27 C-3,28 C‑4,29,30 C-5,31 or C-632,33 in a pyranoid ring, are common. There are fewer reports of simple reduction when the halogen atom is attached to a furanoid ring (C‑1,34,35 C‑5,36,37 or C-638 ), but many nucleoside reactions are known where the halogen atom is bonded to C‑2',39,40 C-3',41,42 or C-5' 43,44 in the substrate. (Each reference listed above refers to a simple-reduction reaction representative of those taking place at the indicated carbon atom.)
.png?revision=1&size=bestfit&width=275&height=206)
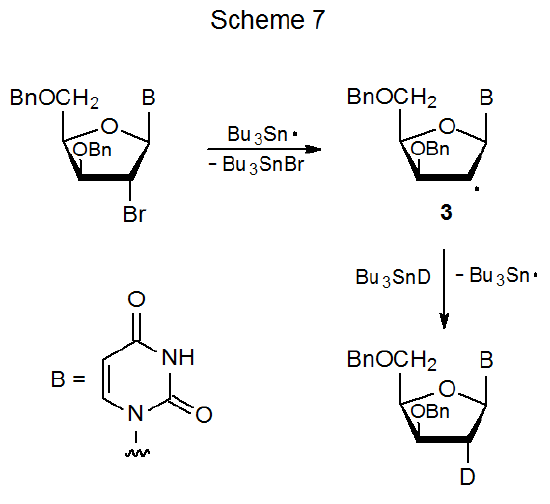
When a halogen atom is replaced by a deuterium atom, simple reduction can provide information about reaction stereoselectivity; thus, in the reaction shown in Scheme 7 the approach of Bu3SnD to the nucleoside radical 3 is from the less hindered face of the furanoid ring.45 Reaction at the 2'‑position in other halogenated nucleosides also is stereoselective, but this selectivity, which is heavily influenced by the structure and positioning of ring substituents, sometimes is modest.46–48
4. Protecting Group Modification
Simple reduction sometimes is used to modify protecting group reactivity.49–57 In the reaction shown in eq 9, for example, replacement of the three chlorine atoms in the trichloroethylidene group with three hydrogen atoms makes hydrolytic removal of this group much easier.49 In a related example, the trichloroacetamido group is converted to the more easily hydrolyzed acetamido group by three, successive simple reduction reactions (eq 10).56 Reaction with tri-n-butyltin hydride as a means of replacing all the chlorine atoms in a protecting group with hydrogen atoms is not always successful because complete dechlorination sometimes is elusive (eq 1158).58–60
.png?revision=1&size=bestfit&width=420&height=170)
.png?revision=1&size=bestfit&width=315&height=233)
.png?revision=1&size=bestfit&width=430&height=216)
5. Acyloxy Group Migration
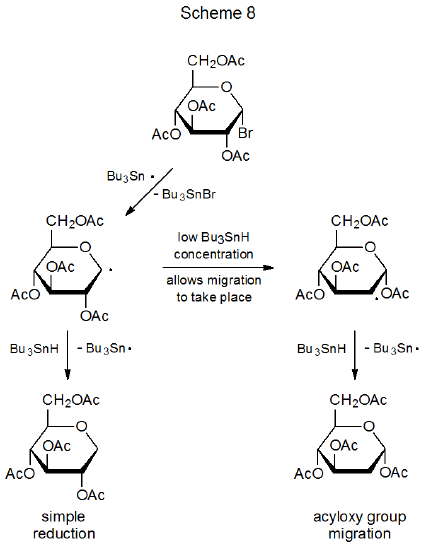
When an anomeric halide with a neighboring acyloxy group reacts with tri-n-butyltin hydride, acyloxy group migration leading to a 2-deoxy sugar competes with simple reduction (Scheme 8).61 If 2-deoxy sugar synthesis (and not simple reduction) is the goal of the reaction,61–64 conditions that maximize group migration need to be selected. Generally, this means maintaining the tri-n-butyltin hydride concentration at a low enough level to allow time for migration to occur.
B. Addition
Halogenated compounds are common radical precursors for addition reactions involving carbohydrates, but other carbohydrate derivatives also function in this capacity; consequently, radical addition reactions are mentioned in most of the chapters in this book. They are discussed extensively not only in this chapter but also in Chapters 10, 12, 13, and 18. In Chapter 18 radical addition is considered from a different point of view, that is, with a focus on the compound to which addition is occurring rather than on the radical precursor.
Addition reactions can be divided into three groups based on what happens after formation of the adduct radical. The first group (addition-abstraction reactions) contains those reactions where the adduct radical abstracts a group or atom from a donor present in solution. Atom abstraction is more common that group abstraction and nearly always involves a hydrogen atom. The second type of reaction (addition-combination) is one in which the adduct radical combines with another radical or an organometallic reagent present in the reaction mixture. In the third reaction type (addition-elimination or addition-fragmentation) the adduct radical forms an unsaturated compound by a β-fragmentation process that eliminates a radical. Examples in which halogenated carbohydrates serve as radical precursors for each of these reaction types are given in the following three sections.
1. Addition-Abstraction Reactions
Formation of a carbon-centered radical by reaction of a glycosyl halide with a tin- or silicon-centered radical is the first step in many addition reactions. An addition-abstraction reaction takes place when a radical produced by halogen-atom abstraction adds to a multiple bond to generate an adduct radical that then abstracts a hydrogen atom. The hydrogen-atom transfer is nearly always a tin- or silicon hydride. An example of this type of reaction is given in eq 12, where a pyranos-1-yl radical generated from the D-mannopyranosyl bromide 4 adds to the α,β-unsaturated ketone 5.65 Pyranos-1-yl radicals generated from halogenated carbohydrates are known to add to α,β-unsaturated nitriles,66,67 esters,67,68 aldehydes,67 ketones,69,70 lactones,71,72 and phosphonates.73,74 In all of these reactions formation of the adduct radical is followed by hydrogen-atom abstraction. Pyranos-1-yl and other carbohydrate radicals formed from halogenated compounds also add to oximes75–77 and electron-deficient enol ethers.78,79 The reactions shown in equations 13 and 14 underscore a critical feature of radical addition reactions, namely, that unless a multiple bond is electron-deficient (eq 13), addition of a nucleophilic radical (which nearly all carbohydrate radicals are) will be too slow to compete effectively with simple reduction (eq 14).80
.png?revision=1&size=bestfit&width=445&height=142)
.png?revision=1&size=bestfit&width=400&height=151)
.png?revision=2&size=bestfit&width=390&height=127)
Addition-abstraction reactions that do not involve pyranos-1-yl radicals are much less common than those that do. An example of a non-pyranos-1-yl radical reaction is shown in eq 15.81 The pyranos-5-yl radical formed in this reaction has reactivity similar to that of a pyranos-1-yl radical because both are nucleophilic species that are stabilized by a ring oxygen atom. This similarity in reactivity can be seen when comparing the reaction shown in eq 1581 with that in eq 16.82 Several addition-abstraction reactions of radicals centered on C-683–86 are known, but reaction of a radical located on an atom in or attached to a pyranoid ring (other than C-1or C-6) is rare.87 There are reports of addition-abstraction reactions where the radical is centered on a carbon atom that is part of or attached to a furanoid ring.88–90
.png?revision=1&size=bestfit&width=365&height=159)
.png?revision=1&size=bestfit&width=390&height=155)
2. Addition-Combination Reactions
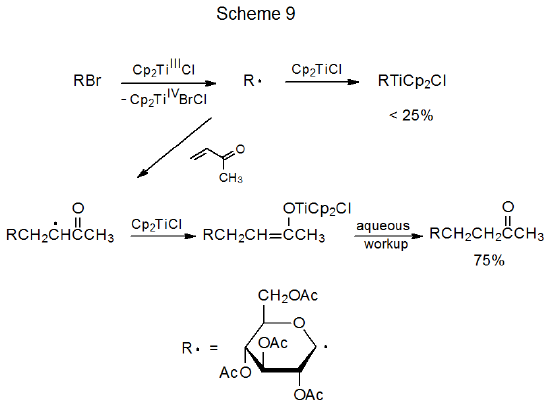
Although radical formation from a glycosyl halide normally involves halogen-atom abstraction by a tin or silicon centered radical, a radical also can be generated by electron transfer to the glycosyl halide from a metal ion, such as the samarium ion in SmI2 or the titanium ion in Cp2TiCl. An example of this type of reaction is shown in Scheme 9, where the pyranos-1-yl radical (R·) is formed by electron transfer from titanium to the glycosyl bromide (RBr).91 In the reaction shown in Scheme 9, radical addition to an electron-deficient multiple bond is faster than combination of the radical with a second molecule of Cp2TiCl. Once addition occurs, however, the situation changes. The reactivity of the adduct radical is so different from the far more nucleophilic pyranos-1-yl radical that the fastest reaction for the adduct radical is combination with a molecule of Cp2TiCl. (Chapters 20-24 contain further discussion of addition-combination reactions brought about by electron-transfer from organometallic complexes to carbohydrate halides.)
3. Addition-Elimination Reactions
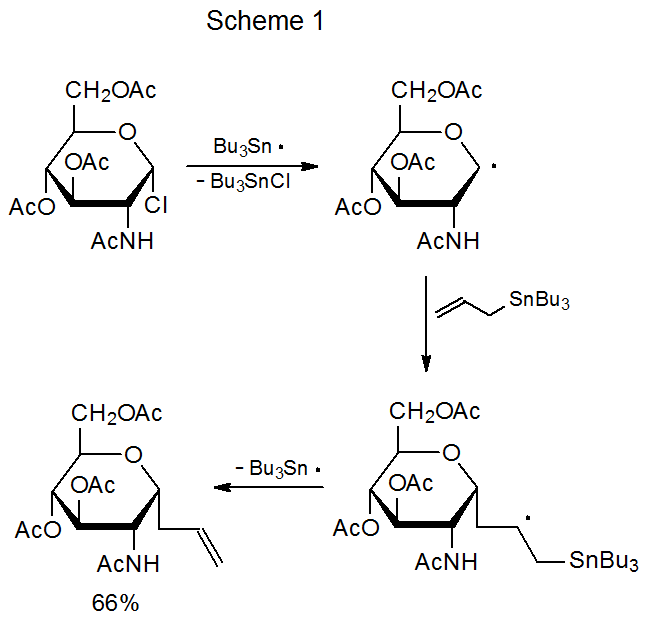
When a carbon-centered radical adds to allyltri-n-butyltin, it begins a sequence of reactions that replaces a halogen atom with an allyl group (Scheme 1).1 This type of transformation (an addition-elimination reaction) often occurs when the halogen atom is attached to C‑11,92–94 but also takes place when such an atom is bonded elsewhere in a carbohydrate framework.95,96 For example, the reaction between the tri-n-butyltin acrylate 6 and the deoxyiodo sugar 7 involves a radical centered at C-6 (eq 17).97
{kind=link}
.png?revision=1&size=bestfit&width=375&height=111)
A reaction mechanistically similar to that shown in eq 17, but one with a quite different outcome, occurs when a halogen atom is abstracted by Bu3Sn· in the presence of t-butyl isocyanide. When this happens, an addition-elimination reaction produces a nitrile (Scheme 10).98
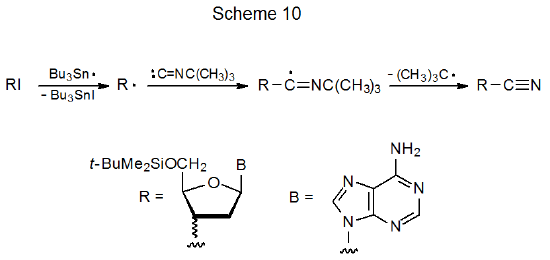
Most addition-elimination reactions transfer an allyl or substituted allyl group from a tin-containing compound to a carbon-centered radical; however, this transfer can be tin-free.99,100 In the reaction shown in eq 18 the allyltin reactant is replaced by allyl ethyl sulfone.99
.png?revision=1&size=bestfit&width=375&height=102)
C. Cyclization
New ring formation is pervasive in the radical reactions of carbohydrates; consequently, radical cyclization, like radical addition, is mentioned in many of the chapters in this book. Significant discussion exists in this chapter because halogenated carbohydrates often are the precursors for radical-based formation of new ring systems. A large portion of Chapter 12 also is devoted to radical cyclization because O-thiocarbonyl compounds frequently are precursors for radicals involved in new ring formation. Chapter 19 is devoted entirely to cyclization reactions and is concerned less with radical formation and more with the internal radical addition that produces new rings.
1. Substrate Reactivity
As with simple reduction reactions, the abstracting radical that begins radical cyclization nearly always is derived from a tin or silicon hydride. The well-established order of reactivity for halogenated compounds with silicon and tin hydrides (RI > RBr > RCl >> RF), mentioned in Section II.B, accounts for the usual selection of a carbohydrate iodide or bromide as the substrate in a cyclization reaction.
Halogen identity is especially critical to dehalogenation with either SmI2 or Cp2TiCl. In the cyclization reaction shown in Scheme 11 the iodide gives a good yield of the substituted cyclopentane, but the bromide is enough less reactive that it produces only bromine-containing dimers arising from reduction of the double bond by SmI2.101 Reaction of a similar compound with Cp2TiCl results in an 82% yield of substituted cyclopentanes from the iodide but only an 18% yield from the corresponding bromide (Scheme 12).102 More forcing reaction conditions might have improved product yield from the bromide because its attempted cyclization returned primarily unreacted starting material.
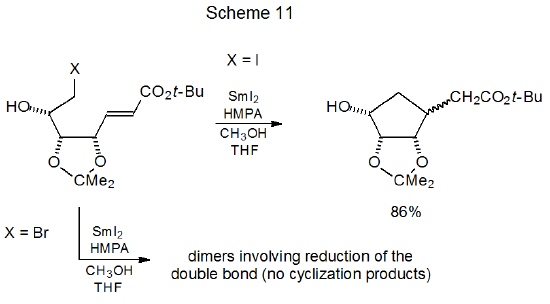
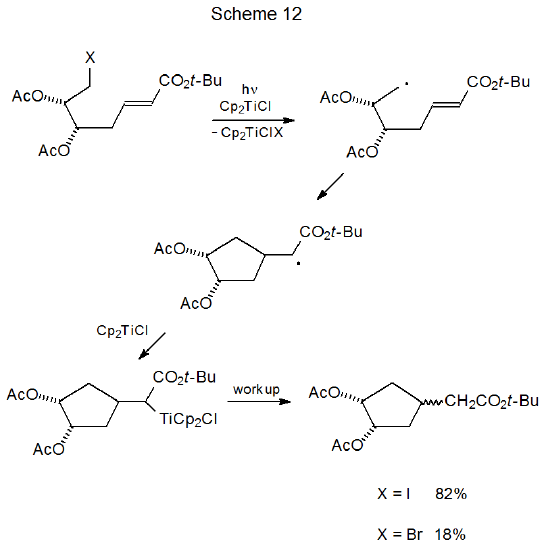
Successful cyclization of halogenated carbohydrates by reaction with SmI2 or Cp2TiCl depends upon reaction conditions and additives. The presence of hexamethylphosphoramide (HMPA) is so important to the reactivity of SmI2 that the cyclic product shown in Scheme 11 does not form unless HMPA is present in the reaction mixture.101 In a similar fashion, UV radiation is critical to the reaction shown in Scheme 12. If it is omitted, little reaction takes place.102
2. Competition between Cyclization and Hydrogen-Atom Abstraction
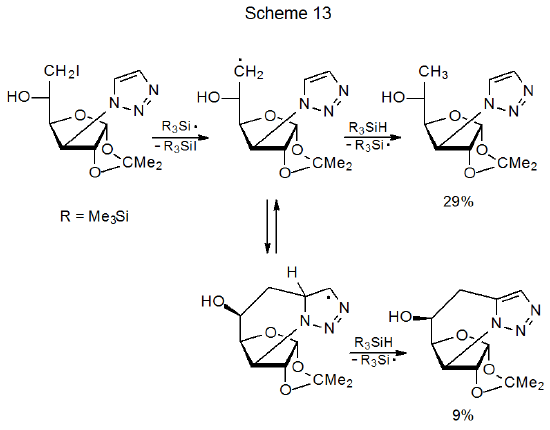
Whenever a cyclization reaction is conducted in the presence of a tin or silicon hydride, hydrogen-atom abstraction by the initially formed radical, leading to simple reduction, competes with cyclization (Scheme 13).38 Other reagents that are not hydrogen-atom transfers but are capable of generating radicals from halogenated carbohydrates (e.g., Bu3SnSnBu3,103 SmI2,101,104,105 and Cp2TiCl102) have the advantage that simple reduction is suppressed. Even though simple reduction is not a problem when using SmI2 and Cp2TiCl, each of these compounds can prevent cyclization by combining with a carbon-centered radical before ring formation takes place; therefore, rapid ring closure remains an important criterion for successful reaction.
3. Organization of Cyclization Reactions
Successful radical cyclization requires a multiple bond and radical center that are suitably positioned with respect to each other. The radical center in such a reaction can be either on an atom that is part of the carbohydrate framework (Figure 2) or on a substituent group. The same possibilities exist for the multiple bond. For purposes of discussion it is useful to divide cyclization reactions into the four basic types shown in Figure 3. This Figure also contains a short-hand terminology that has been proposed to identify each reaction type.105 Chapter 19 contains extensive tables of cyclization reactions in which radicals are formed from halogenated and nonhalogenated carbohydrates.
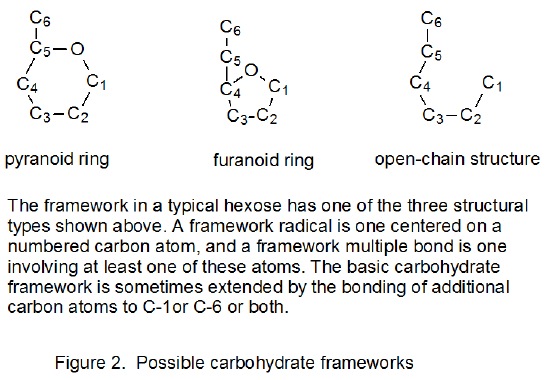
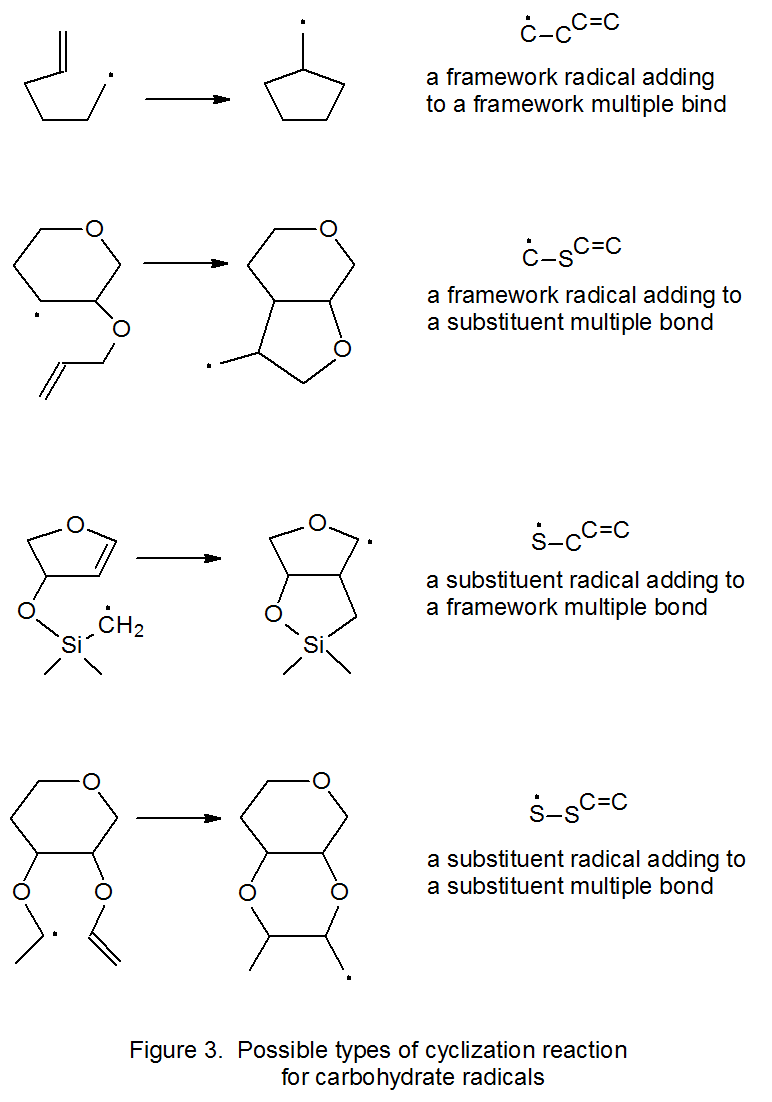
a. Addition of a Framework Radical to a Framework Multiple Bond
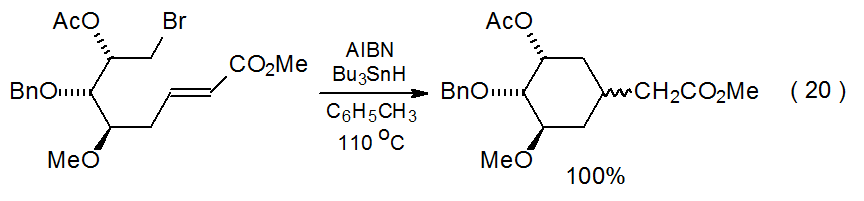
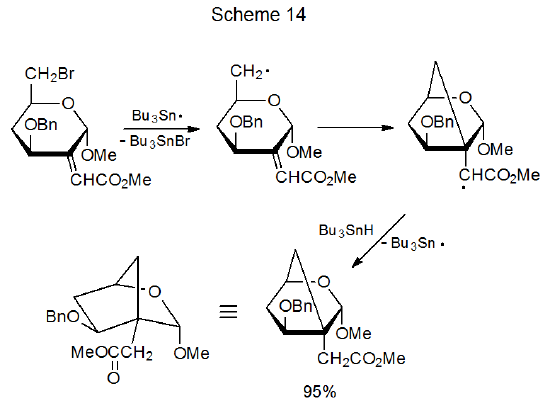
The possibilities for a framework radical adding internally to a framework multiple bond are limited by the size of the rings that are easily produced; thus, only five- and six-membered rings generally form rapidly enough to compete with other radical reactions. Since radical cyclization produces five-membered rings more quickly than six-membered ones, the typical cyclization reaction produces a pair of heavily substituted, cyclopentane derivatives (eq 19106).106–111 When five-membered ring formation is not possible but producing a six-membered ring is, cyclization gives a substituted, cyclohexane derivative (eq 20).112 Addition of a framework radical to a framework multiple bond also can produce a bicyclic compound (Scheme 14).113
.png?revision=1&size=bestfit&width=410&height=107)
.png?revision=1&size=bestfit&width=435&height=101)
Although forming either a five- or six-membered ring is the typical result of radical cyclization, larger rings are possible if the carbohydrate framework is held so that the radical center easily can approach the multiple bond. Striking examples of such a situation are found in the reactions shown in equations 21 and 22, where the O-isopropylidene groups cause the iodides 8 and 9 to adopt conformations that favor formation of seven- and eight-membered rings, respectively.114
.png?revision=1&size=bestfit&width=425&height=155)
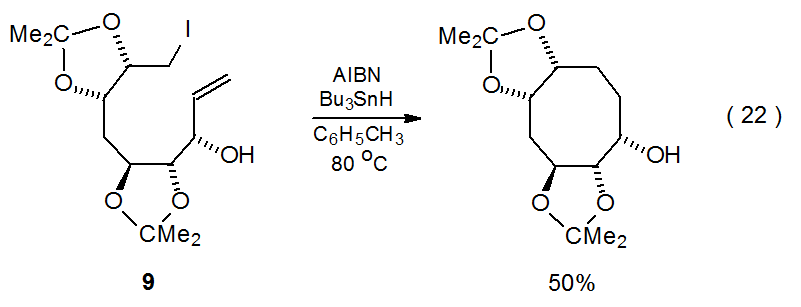.png?revision=1&size=bestfit&width=400&height=153)
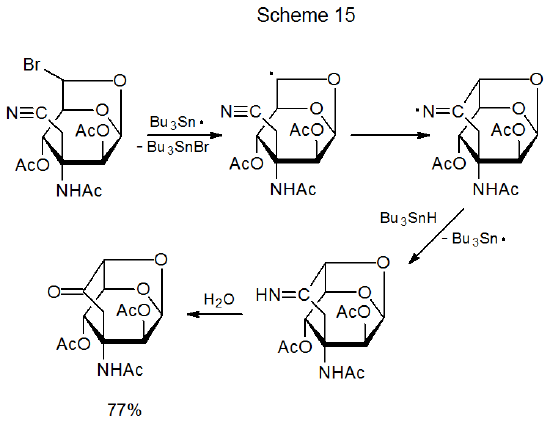
One way to increase the reactivity of a multiple bond in a radical cyclization reaction is to replace one of the carbon atoms with an electronegative heteroatom.115–121 The remaining carbon atom then will have electron density drawn from it and, as a result, have enhanced reactivity toward nucleophilic radicals. The oxime 10 contains a double bond activated in this way (eq 23).115 Having reactive centers able easily to come within bonding distance translates into cyclic products being formed in reactions involving other carbon–nitrogen116–119 and even carbon–oxygen120,121 multiple bonds. In the reaction shown in Scheme 15, for example, ring formation occurs because the radical center easily comes into contact with the cyano group.118
.png?revision=1&size=bestfit&width=380&height=129)
b. Addition of a Framework Radical to a Substituent Multiple Bond
An example of a reaction in which a framework radical adds to a substituent multiple bond to form a five-membered ring is shown in eq 24.122 If five-membered-ring formation introduces too much strain into a system, reaction to give a larger ring becomes a possibility; thus, the radical centered at C-5 in 11 adds to the substituent double bond to form a six-membered ring (Scheme 16) and, in so doing, avoids producing highly strained, trans-fused, five-membered rings.123
.png?revision=1&size=bestfit&width=410&height=129)
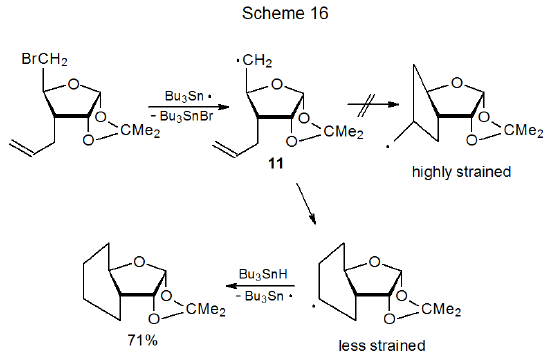
Although bimolecular addition of a carbohydrate radical to a multiple bond is fast enough to be observed only when the multiple bond is electron-deficient, radical cyclization is not limited in this way. Having a radical center, such as the one at C-5 in 11 (Scheme 16), in close proximity to a double bond makes cyclization competitive with other radical reactions (e.g., simple reduction) even though the multiple bond is not electron-deficient.
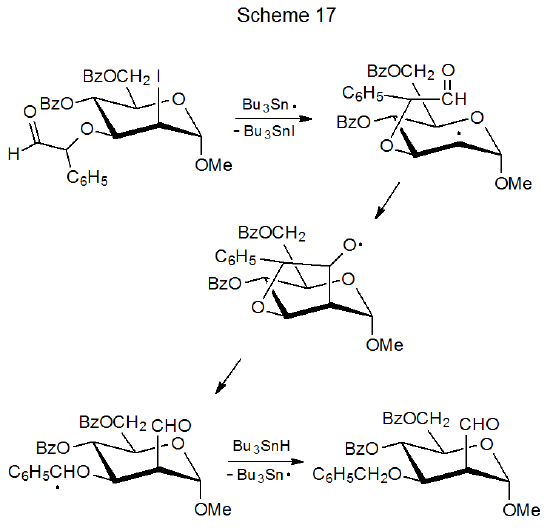
Radical cyclization followed by ring opening that breaks the newly formed bond is a degenerate addition-elimination reaction that is rarely useful or even detectable. Sometimes, however, ring opening breaks a different bond from that produced during cyclization.124,125 The result of such a reaction is an addition-elimination process, such as that shown in Scheme17, where the effect of the reaction is migration of a part of a substituent group.124
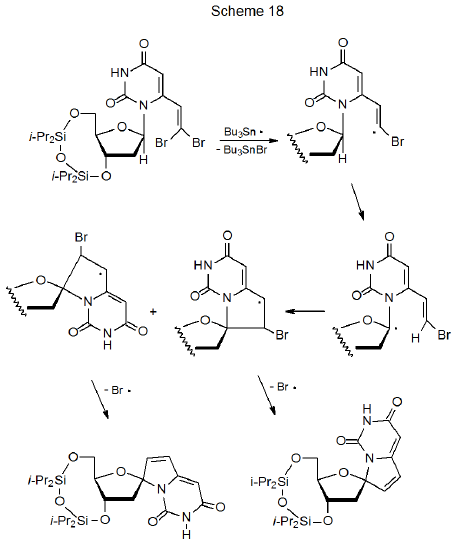
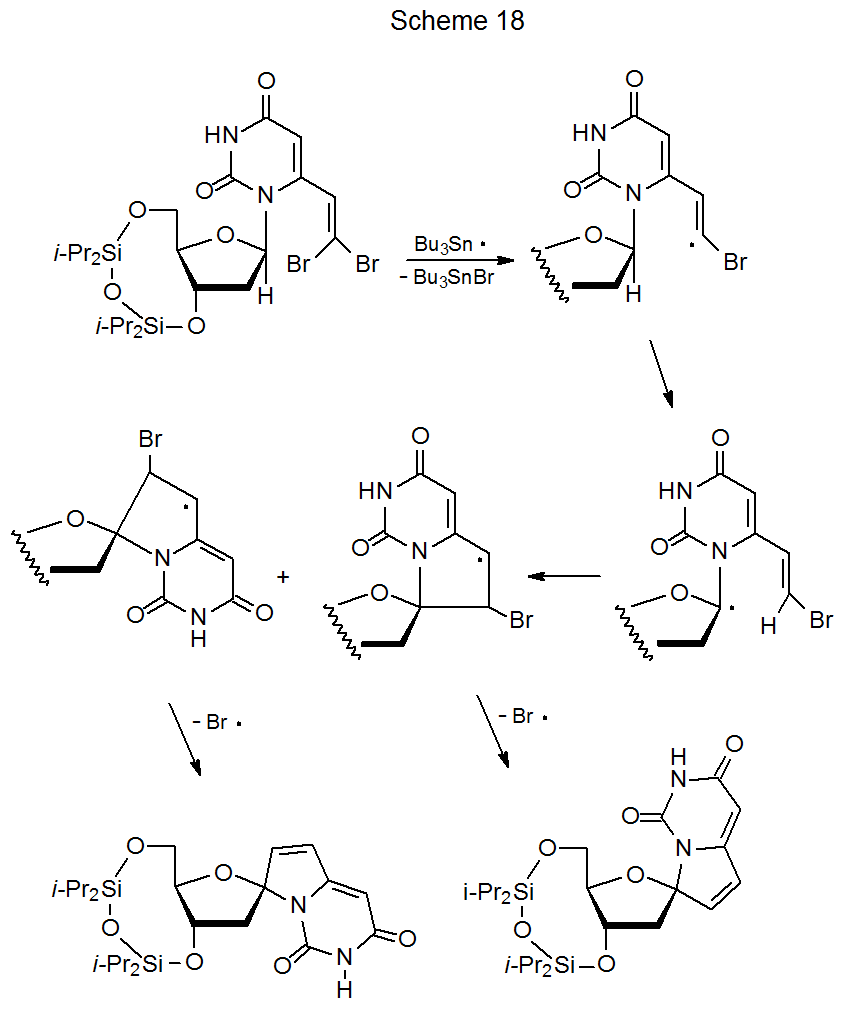
A unique type of cyclization between a framework radical and a substituent multiple bond takes place in nucleosides that have properly placed dibromovinyl groups.103,126–131 Bromine-atom abstraction by Bu3Sn· produces a vinyl radical that begins a sequential reaction leading to two spiro compounds (Scheme 18).103 Using Bu3SnSnBu3 in this reaction (rather than Bu3SnH) improves the yield of the cyclization product because competing simple reduction involving a tin hydride is not an option.103
c. Addition of a Substituent Radical to a Framework Multiple Bond
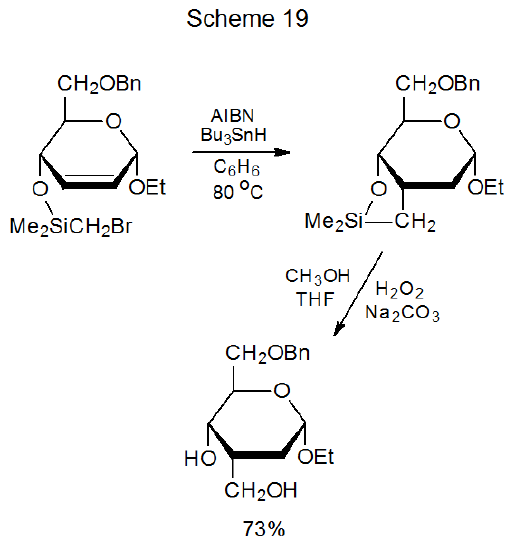
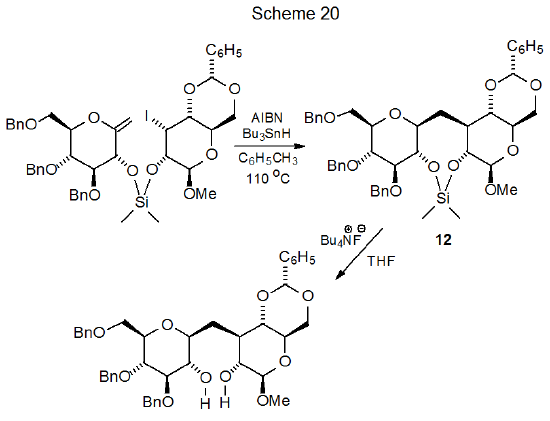
Cyclization involving halogenated carbohydrates often occurs when the halogen atom is part of a substituent group and the multiple bond is located in the carbohydrate framework. The substrate in many of these reactions is a silyl ether with a bromine atom incorporated into the silicon-containing group.132–139 An example is shown in Scheme 19, which describes a reaction that stereoselectively creates a new C–C bond at C-3 in the product.132 Nonradical reaction of the resulting product transforms it into a diol (Scheme 19) that can be readily converted into other compounds. An extension of this type of reaction to a pair of saccharide units connected by a silaketal tether leads to formation of a protected C-disaccharide (12) from which the tether is easily removed (Scheme 20).140
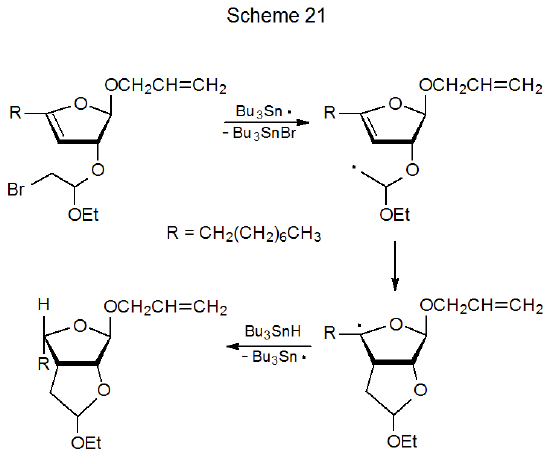
Cyclization of an unsaturated carbohydrate in which an acetal substituent contains a halogen atom is similar to cyclization of brominated silyl ethers such as that pictured in Scheme 19. High stereoselectivity is the norm in these reactions.141–147 In the process shown in Scheme 21, for example, the stereochemistry at C-3 is controlled by the kinetically and thermodynamically favored formation of a cis-fused ring system.141 [There is a second chiral center (C-4) created during this reaction. The stereochemistry at this center is determined by the least-hindered approach of tri-n-butyltin hydride to the reacting radical.] The reaction shown in eq 25 provides an example of new ring formation when a radical formed from a halogen-containing, acetal substituent adds internally to a triple bond.142
.png?revision=1&size=bestfit&width=355&height=104)
d. Addition of a Substituent Radical to a Substituent Multiple Bond
Sometimes in a radical cyclization reaction neither the carbon atom bearing the radical center nor the carbon atoms of the multiple bond are part of the carbohydrate framework.148,149 When this occurs, the carbohydrate portion of the molecule has only an indirect influence on the reaction; that is, it can affect reaction stereoselectivity by acting as a chiral auxiliary, or its cyclic structure can bring reactive centers into bonding distance. It is the latter role that assists eleven-membered ring formation in the reaction shown in eq 26.148
.png?revision=1&size=bestfit&width=420&height=263)
D. Elimination
Free-radical elimination takes place when a vicinal dihalide reacts with tri-n-butyltin hydride (eq 27).150 A similar reaction occurs if one of the vicinal substituents is a halogen atom and the other contains an O-thiocarbonyl group.151,152 In the reaction shown in eq 28151 there is little doubt that the initial interaction between Bu3Sn· and compound 13 in most instances is with the substituent at C-3' because the rate constant for reaction of Bu3Sn· with a compound containing an O-thiocarbonyl group is much larger than that for reaction with a chlorinated compound.
.png?revision=1&size=bestfit&width=320&height=182)
.png?revision=1&size=bestfit&width=335&height=184)
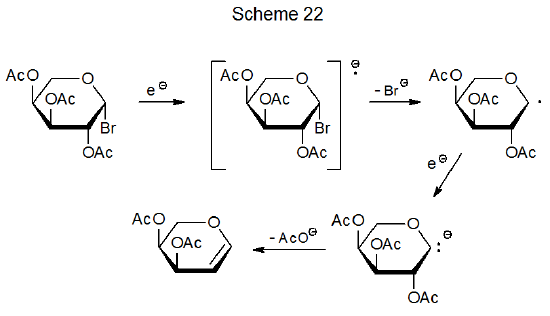
Electrochemical reduction of glycosyl halides (and their sulfur-containing analogs154a) begins with electron transfer to the halide to produce a radical anion that reacts further to give the corresponding glycal (Scheme 22153). Reaction of glycosyl bromides with zinc dust also leads to glycal formation by electron-transfer to a glycosyl halide (eq 29).154b The mechanism for this reaction may parallel that shown in Scheme 22, but the possibility also exists that an organozinc intermediate forms following the initial electron transfer.
.png?revision=1&size=bestfit&width=325&height=104)
E. Ring-Opening
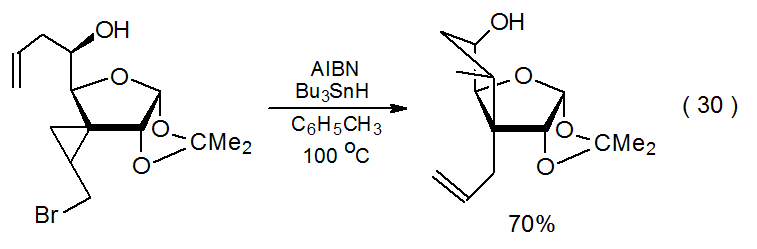
If halogen-atom abstraction produces a cyclopropylcarbinyl radical, cyclopropane ring opening takes place. The radical remaining after ring opening then can undergo hydrogen-atom abstraction,155 addition,156 or, as shown in eq 30, cyclization.157
.png?revision=1&size=bestfit&width=380&height=125)
F. Internal Hydrogen-Atom Abstraction
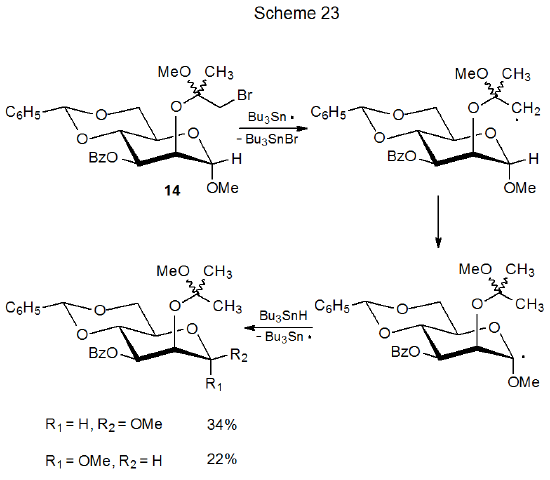
Reaction of a halogenated carbohydrate to form a carbon-centered radical rarely results in this radical abstracting a hydrogen atom from a carbon–hydrogen bond in another molecule because such abstraction is, in most instances, too slow to compete with other radical reactions. If, however, the abstraction is internal and the radical is quite reactive (primary, vinyl or aryl), hydrogen-atom abstraction can take place.158–160 Dehalogenation of the bromide 14 begins such a reaction by producing a primary radical that abstracts a hydrogen atom from C‑1, a process that leads to epimerization at this carbon atom (Scheme 23).158 Internal hydrogen-atom abstraction by a vinyl radical takes place in the reaction pictured in Scheme 18.103
{kind=link}
G. Radical Combination
1. Dimerization
Radical combination is not a common synthetic reaction for carbohydrates, but it does take place when conditions are adjusted so that reactions such as hydrogen-atom abstraction are to slow to compete.161–163 Having the source of the chain-carrying radical be Me3SnSnMe3 (as opposed to a tin hydride) reduces the rate of hydrogen-atom abstraction by the carbohydrate radical to the point that the three dimers shown in eq 31 are formed. {Similar dimer formation takes place during reaction of glycosyl phenyl sulfones [Chapter 3, Section VII.B.1.c] and glycosyl phenyl selenides [Chapter 4, Section II.B.6]} Electrochemical reactions of glycosyl halides also produce radical dimers.163
.png?revision=1&size=bestfit&width=375&height=154)
2. Reaction with Molecular Oxygen
Reaction of halogenated carbohydrates with tri-n-butyltin hydride in the presence of oxygen replaces the halogen atom with a hydroxyl group.164–168 Since combination of a carbon-centered radical with molecular oxygen is either diffusion-controlled or nearly so (k \(\cong\) 2 x 109 M‑1s-1),169 any other reaction of the radical taking place in the presence of oxygen must be rapid enough to happen before O2 reaches the radical center. An example of a cyclization reaction that is fast enough to meet this criterion is shown in eq 32.164 Since the oxygen concentration in the reaction mixture is approximately 1 x 10-3 M, the rate constant for cyclization needs to be at least 1 x 107 s-1 in order for an acceptable yield of a cyclic product to be obtained.166b A proposed mechanism for replacement of a halogen atom with a hydroxyl group is given in Scheme 24.170
.png?revision=1&size=bestfit&width=340&height=168)
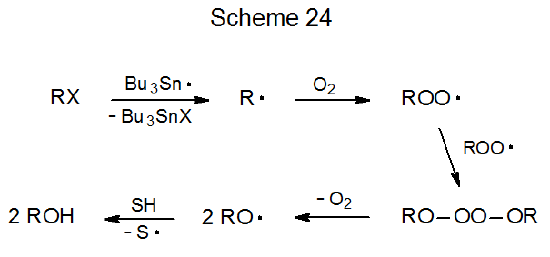
Other reagents and reaction conditions also cause replacement of halogen atoms with hydroxyl groups. These include Et3B–O2 initiated reaction of a deoxyiodo sugar with molecular oxygen171,172 and adding O2 to a reaction mixture in which AIBN is both initiator and reactant.166 Another set of conditions leading to replacement of a halogen atom with a hydroxyl group consists of reacting a deoxyiodo sugar with NaBH4 and O2 in the presence of a catalytic amount of Co(salen) (eq 33173).162,173–175 A comparative study found Co(salen)-catalyzed oxidation reactions to be experimentally more convenient than those with a tin hydride.162
.png?revision=1&size=bestfit&width=340&height=238)
H. Water-Soluble Halides
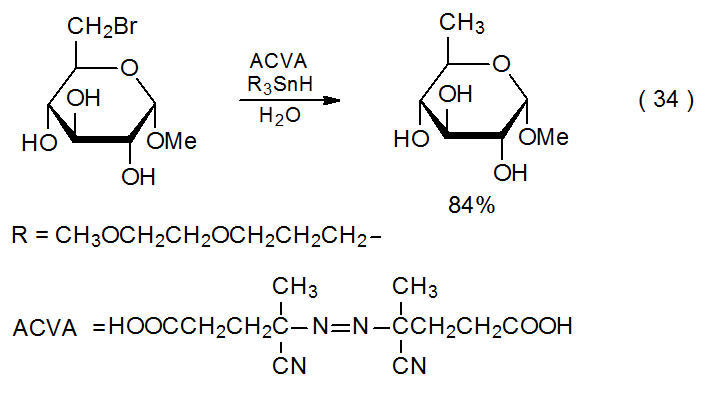
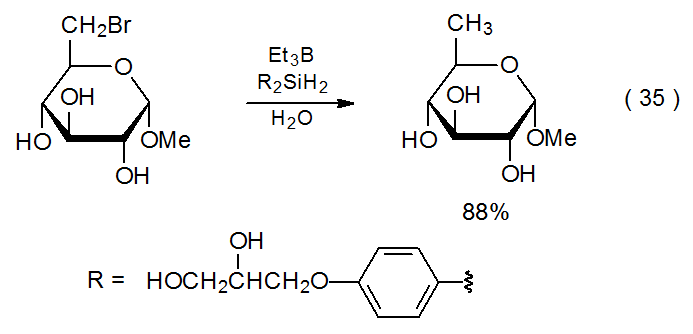
Most halogenated carbohydrates used in synthesis are rendered soluble in organic solvents by the introduction of various hydroxyl protecting groups, but sometimes it is useful to be able to conduct dehalogenation reactions in aqueous solution on unprotected or partially protected carbohydrates. Such a situation requires a water-soluble replacement for the tin and silicon hydrides typically used. Water-soluble hydrogen-atom transfers can be formed by replacing the alkyl substituents normally attached to tin or silicon with more polar ones. This replacement produces hydrides that are sufficiently soluble in water to allow simple reduction to take place in aqueous solution (eq 34176 and eq 35177).
.png?revision=1&size=bestfit&width=355&height=198)
.png?revision=1&size=bestfit&width=350&height=169)
There are potential advantages to conducting reactions in water, advantages that extend beyond substrate solubility.176,177 One of these is that reactions conducted in aqueous solution may exhibit new reactivity because, rather than taking place in an essentially nonpolar liquid, these reactions occur in a polar, heavily hydrogen-bonded solvent. Since water generally does not participate in radical reactions, it is effectively an inert solvent. Also, using water as the reaction medium can reduce or even eliminate the need for recycling or disposal of organic solvents.
I. Hypohalites
Hypohalites are intermediates in the formation of alkoxy radicals. Reactions of hypohalites and the alkoxy radicals they produce are discussed in Chapter 6.
J. Organotin Hydrides
The majority of reactions of halogenated carbohydrates use an organotin hydride (nearly always Bu3SnH) as a hydrogen-atom transfer and as a source of a chain-carrying-radical; however, there are serious problems associated with the toxicity of tin-containing compounds and the difficulty in removing their residues from reaction products. A variety of solutions to these problems have been proposed. Since most of these solutions apply not only to reactions of halogenated carbohydrates but also to those of a broad range of carbohydrate derivatives, the solutions will not be discussed here (and then repeated in later chapters); rather, they are gathered together in Appendix 1, which is found at the end of this book.