
V. Examples of Radical Philicity in Reactions of Carbohydrates
- Page ID
- 23938
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)A. Hydrogen-atom abstraction
The reactions shown in equations 4 and 5 illustrate the importance of radical philicity in hydrogen-abstraction reactions by showing that the nucleophilic radical R· abstracts the electron-deficient hydrogen atom attached to sulfur (eq 5) more rapidly than the electron-rich hydrogen atom bonded to tin (eq 4).33–35 The differences in rate constants and enthalpies for these two reactions underscore the fact that radical philicity affects the stability of the transition-state structure in a reaction but not the overall energy changes due to bond breaking and bond formation.
.png?revision=1){kind=link}
.png?revision=1){kind=link}
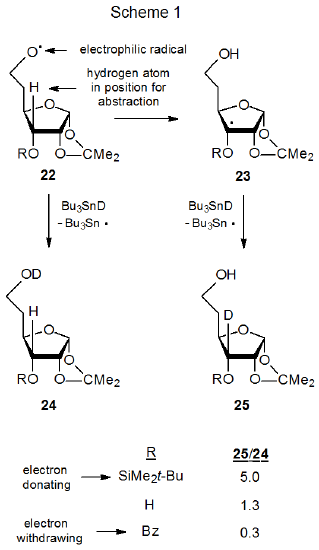
Comparing the three reactions pictured in Scheme 1 draws attention to the effect of radical philicity on hydrogen-atom abstraction from carbohydrates. In each of these reactions the oxygen-centered radical 22 either abstracts a deuterium atom from Bu3SnD to give the deuterated alcohol 24, or it reacts internally with H-3 to generate the carbon-centered radical 23.36 After the radical 23 forms, it then abstracts a deuterium atom for Bu3SnD to give the second reaction product (25). The relative amounts of products 24 and 25 provide a measure of external (deuterium) versus internal (hydrogen) abstraction. As H‑3 becomes less electron rich, internal reaction (22\(\rightarrow\)23) becomes less competitive. External abstraction (22\(\rightarrow\)24), on the other hand, should not be noticeably affected by changes in substituents at C-3. If, as expected, the transition states for the internal hydrogen-abstraction reactions shown in Scheme 1 are early, these reactions support the idea that polarity matching has a critical role in determining the favored reaction pathway for the radical 22.
B. Radical Addition
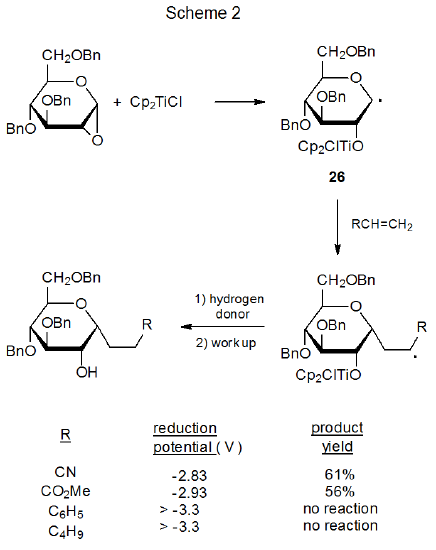
Pyranos-1-yl radicals add readily to electron-deficient, carbon-carbon double bonds but are much less reactive toward double bonds lacking electron-withdrawing substituents.37,38 A group of reactions that illustrates this difference in reactivity is found in Scheme 2.37 The ability of the pyranos-1-yl radical 26 to add to the unsaturated compounds shown in Scheme 2 correlates with the reduction potentials of these compounds; that is, addition to compounds with less negative reduction potentials occurs more rapidly than addition to compounds with more negative reduction potentials.37
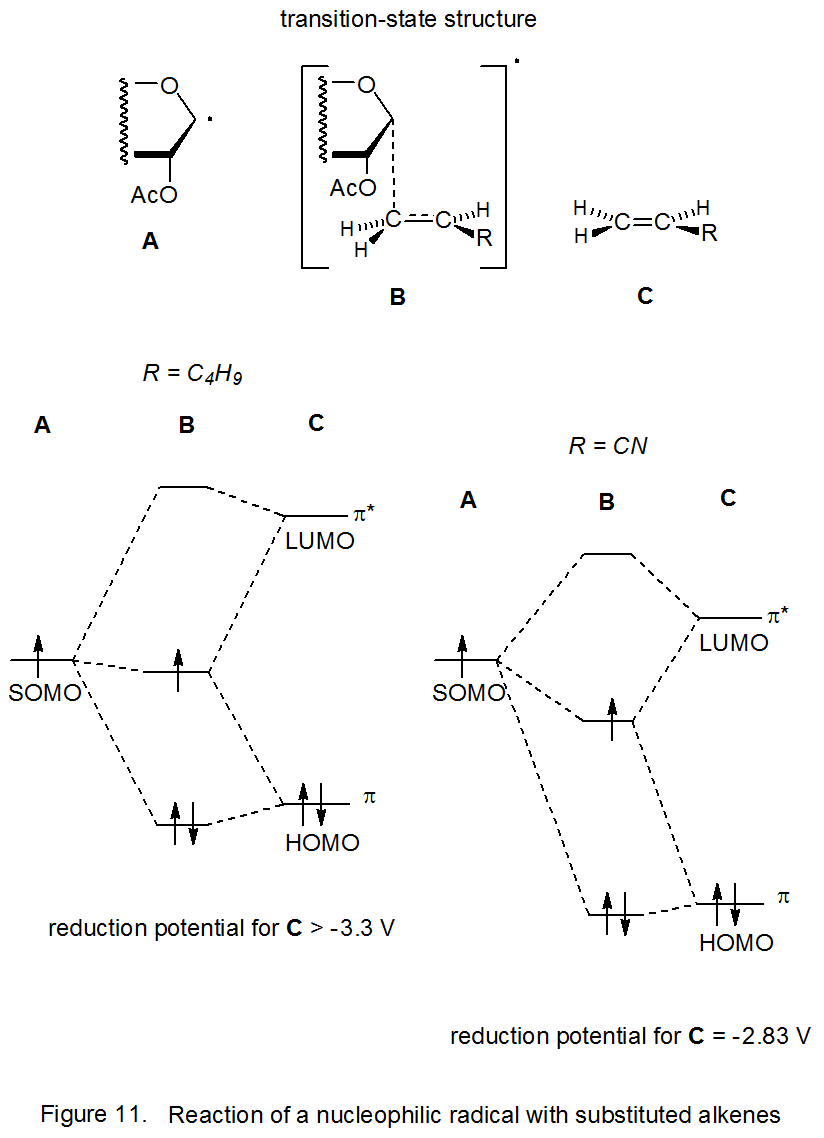
Since the reduction potential in a substituted alkene is a measure of the ease of introducing an electron into a π* orbital, this potential becomes an indicator of energy-level positioning. When comparing two reduction potentials, the less negative one has a lower energy level for the π* orbital. Because this lower energy level causes the π* orbital (LUMO) to interact more effectively with the SOMO of the adding radical, transition-state stabilization due to frontier-orbital interaction increases as the reduction potential for the substituted alkene becomes less negative (Figure 11).
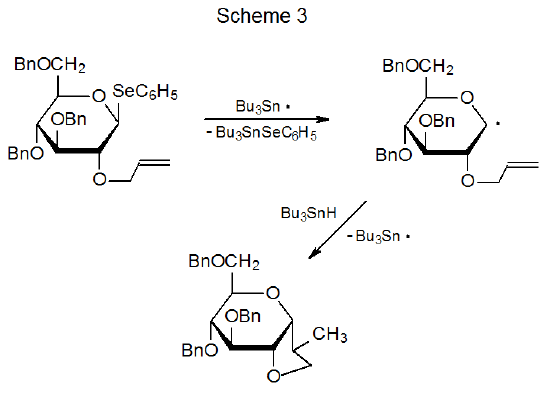
Although the addition of a nucleophilic radical to a π bond that is not electron-deficient is too slow to be observed in the reactions shown in Scheme 2, the situation changes when reactions become intramolecular. For π bonds that are 1,5- or 1,6-related to a radical center, intramolecular addition can take place even if the π bond is not decidedly electron-deficient (Scheme 3).39 As far as overall reaction rate is concerned, forced, close proximity of the radical center to the π bond can compensate for a small transition-state stabilization caused by a large separation in energy levels of interacting, frontier orbitals.
{kind=link}
C. Polarity-Reversal Catalysis
The philicity of radicals involved in hydrogen-abstraction reactions provides the basis for a phenomenon known as polarity-reversal catalysis.4,33–35,40 This type of catalysis, which is responsible for the effect that thiols have on the reactions of carbohydrate acetals and ethers, is discussed in Section III of Chapter 5 in Volume II.