
IV. Explaining Radical Philicity
- Page ID
- 23937
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)A. Valence Bond Theory
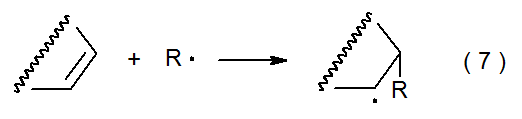
One way to explain the existence of radical philicity begins with the addition reaction shown in eq 7. The transition-state structure in this reaction can be represented as a hybrid of valance-bond structures. If there is no separation of charge, the transition state can be represented by the contributors 3 and 4 shown in Figure 1. Unequal electron distribution at the transition state can be taken into account by including additional resonance contributors. If in the transition state there is a transfer of electron density from R· to the C–C double bond, this transfer can be represented by adding contributors 5 and 6 (Figure 2). If electron transfer is in the other direction, resonance contributors 7 and 8 (but not 5 and 6) make a significant contribution to the transition state structure (Figure 2).
.png?revision=1&size=bestfit&width=265&height=60)


1. Nucleophilic Radicals
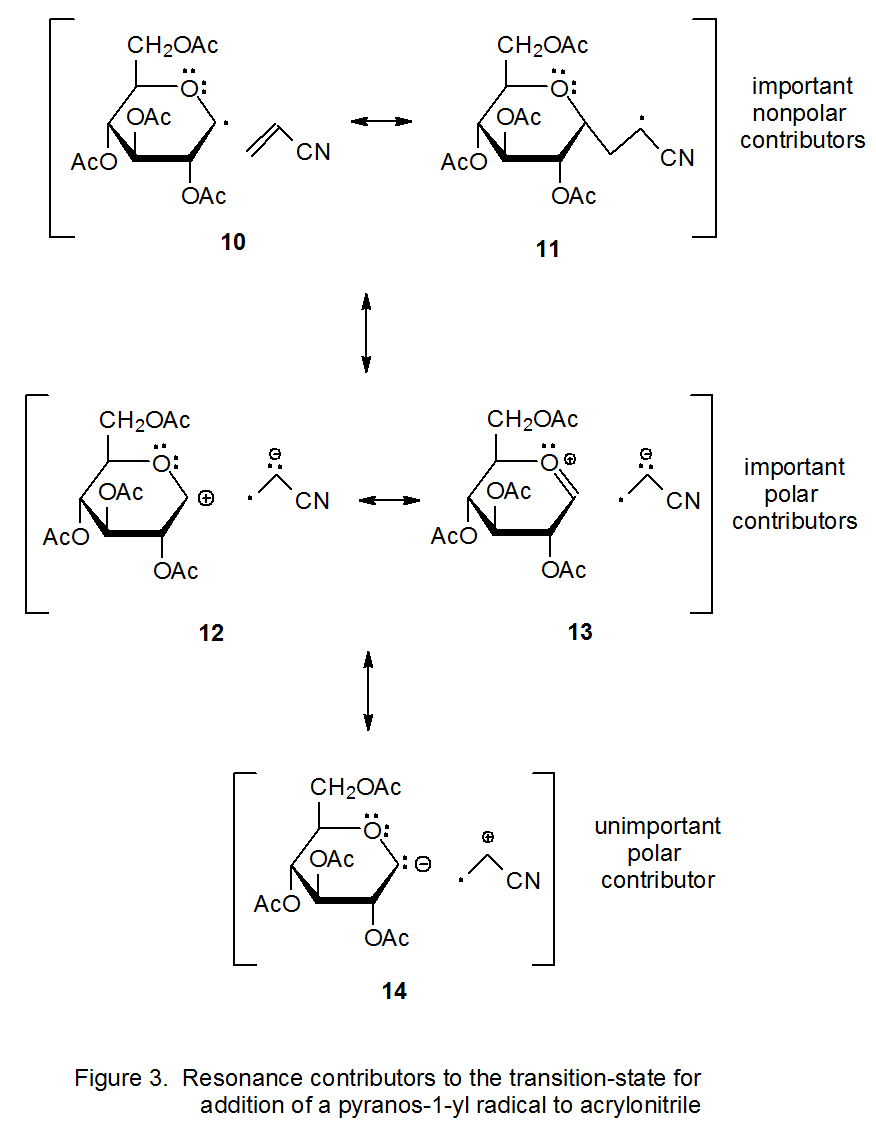
To illustrate the way in which valance bond theory explains radical nucleophilicity, it is instructive to examine the reaction shown in eq 8. In this reaction the D-glucopyranos-1-yl radical 9 is considered to be nucleophilic because it adds to the electron-deficient double bond in acrylonitrile.24 The valence-bond structures 10-14 (Figure 3) all potentially contribute to the transition-state structure in this reaction. Structures 10 and 11 are major contributors that have no separation of charge. Structures 12 and 13 are minor but significant contributors, minor because they involve separation of charge but significant because they stabilize either negative charge (12) or positive and negative (13) charges effectively. Structure 14 is not significant because it has charge-separation and the charges are not effectively stabilized. Since structures 12 and 13 make a greater contribution than does 14 to the transition state in this reaction (eq 8), the radical 9 becomes a net electron-donor at the transition state and, thus, is considered to be nucleophilic.
.png?revision=1&size=bestfit&width=415&height=112)
2. Electrophilic Radicals
If a radical center has a sufficiently strongly electron-withdrawing substituent (or substituents) attached, the inherently nucleophilic character of a carbon-centered radical is reduced to the point that the radical becomes electrophilic.18 For example, the malonyl radical 1, which has two electron-withdrawing groups attached to the radical center, is considered to be electrophilic because it adds to the electron-rich double bond in the D‑glucal 2 (eq 3).7 The electrophilicity of 1 can be understood primarily in terms of the importance to the transition-state structure of the charge-transfer contributor 15, a structure in which the electron-withdrawing methoxycarbonyl groups stabilize the negative charge and the ring oxygen atom stabilizes the positive charge (Figure 4). Together these stabilizing interactions increase the contribution at the transition state from a structure in which the radical 1 is acting as an electrophile by accepting electron density from the D-glucal 2. The resonance structure 16 is not an important contributor at the transition state because within 16 there is a destabilizing shift of electron density away from a radical center that contains electron-withdrawing, methoxycarbonyl substituents.
.png?revision=1){kind=link}

3. Ambiphilic Radicals
Inherent in defining radical philicity in terms of electron-transfer is the idea that the philicity of a radical is a function of the reaction in question. This means that instead of describing a radical as nucleophilic, it should be described as nucleophilic in a particular reaction. It is fair to say, however, that radicals that are moderately or strongly electrophilic or nucleophilic in one reaction are likely to have the same philicity in all reactions, but radicals that are weakly nucleophilic or electrophilic in one reaction are better candidates for a philicity change in a different reaction. Radicals that are nucleophilic in one reaction but electrophilic in another are classified as ambiphilic.
B. Molecular Orbital Theory: Frontier-Orbital Interactions
When a transition-state structure for a reaction resembles the structure of the starting materials, frontier-orbital interactions provide qualitative information about energy changes taking place at the transition state. (Since frontier orbitals are based on the structures of the starting materials, the further the transition state is along the reaction pathway the less reliable frontier-orbital interactions will be in predicting or rationalizing reactivity.) According to Hammond’s postulate,25 an exothermic reaction should have an early transition state with a structure resembling that of the starting materials; therefore, such a reaction should be suitable for analysis by frontier-orbital interactions. A reaction involving addition of a carbon-centered radical to a carbon–carbon double bond is a prime candidate for this type of analysis because such a reaction replaces a π bond with a more stable σ bond, a change that should produce a decidedly exothermic reaction.1,3,6,26
1. Nucleophilic Radicals
A beginning point for explaining radical nucleophilicity in terms of frontier-orbital interactions is found in Figure 5, which pictures the singly occupied molecular orbital (SOMO) in the radical 17 interacting with both the π* (LUMO) and the π (HOMO) orbitals of the alkene 18. Identifying the most important interaction is critical to determining the nucleophilicity of the adding radical. When the SOMO of 17 interacts with the alkene 18, the greater interaction is with the π* orbital of the alkene (Figure 5).6 Convincing evidence supporting this position comes from plotting calculated HOMO and LUMO energies of substituted alkenes against the natural logarithm of the relative rate constants (ln krel) for addition of a carbon-centered radical (the tert-butyl radical was used) to these alkenes.27 A linear correlation exists between ln krel and LUMO energies, but no such correlation exists between ln krel and HOMO energies. The correlation with LUMO energies then is consistent with the dominant frontier-orbital interaction being between the SOMO of the radical 17 and the π* orbital of the alkene 18 (Figure 5).

The next step in understanding how frontier-orbital interactions can explain radical nucleophilicity involves the addition of the radical 17 to the alkene 19, a compound in which the double bond contains the electron-withdrawing substituent Z (Figure 6). When Z replaces one of the hydrogen atoms attached to a doubly bonded carbon atom, the π* orbital is stabilized and the associated energy level moves closer to that of the SOMO of 17.28 This change in energy level position increases the interaction between the SOMO and the π* orbital (Figure 6). Greater interaction translates into a lower transition-state barrier for reaction; therefore, the radical 17 will add more rapidly to the alkene containing the electron-withdrawing Z group than to an unsubstituted alkene. This preferential reaction with electron-deficient alkenes makes the radical 17 nucleophilic.

It is possible to increase the nucleophilicity of a carbon-centered radical still further if its SOMO energy level moves even closer to that of the π* orbital of an alkene. This type of change occurs when an oxygen atom is attached directly to the radical center because interaction between the p‑type orbital on the carbon atom and the p-type orbital on the adjacent oxygen atom raises the SOMO energy level in the resulting radical (20) (Figure 7). This higher energy level places the SOMO closer energetically to the π* orbital of the reactant alkene. Such a change further increases orbital interaction and in so doing causes greater transition-state stabilization. The enhanced reactivity, due to the presence of the attached oxygen atom, means that the radical 20 will be even more nucleophilic than 17; thus, this oxygen-substituted radical (20) is considered to be strongly nucleophilic.

There is an additional way of viewing the frontier-orbital interaction between an alkene and a carbon-centered radical. Understanding this alternative view begins by recalling that the major, frontier-orbital interaction between a carbon-centered radical and an unsaturated compound is between the SOMO of the radical and the π* orbital (LUMO) of the alkene (Figures 5-7). Since SOMO-LUMO interaction is the most important and since any electron donation at the transition state resulting from this interaction must involve electron transfer from the SOMO (the LUMO has no electrons to transfer), the radical is acting as an electron donor and, therefore, is behaving as a nucleophile.29
2. Electrophilic Radicals
If a hydrogen atom attached to a carbon-centered radical is replaced by an electron-withdrawing substituent (e.g., a cyano or carbonyl group), the resulting radical becomes more electrophilic.6,18,30,31 Additional substitution of this type further increases radical electrophilicity (Figure 8). The electron-withdrawing group causes the energy level associated with the singly occupied molecular orbital of the substituted radical to move to a position lower in energy; that is, the radical becomes more stable.6,32 When the energy level of an SOMO in a carbon-centered radical becomes sufficiently low, the major, frontier-orbital interaction with an alkene changes; that is, the primary interaction is no longer with the π* orbital of the alkene but rather with its π orbital (Figure 9). When this change occurs, the primary shift in electron density at the transition state is away from the filled HOMO toward the partially filled SOMO; thus, the radical is electrophilic.

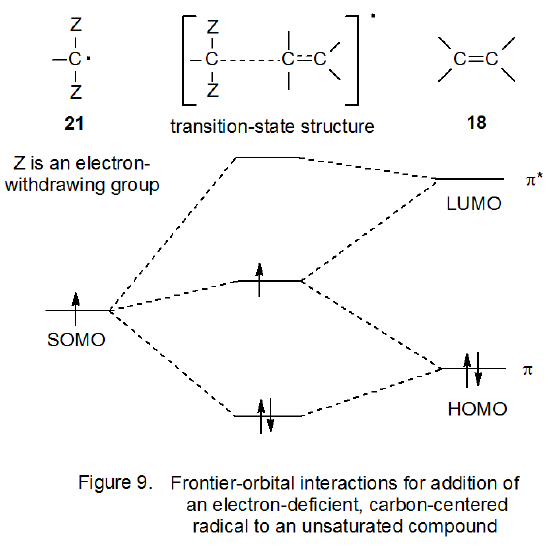
Figure 10 pictures the frontier-orbital interaction of the radical 21 with an alkene that has an electron-donating substituent. Since the HOMO for the substituted alkene (Figure 10) is higher in energy than the HOMO of the unsubstituted alkene (Figure 9),28 transition-state stabilization from SOMO-HOMO interaction will be greater for reaction involving the substituted alkene (Figure 10). Due to this greater stabilization, the radical 21 reacts more rapidly with the more electron-rich alkene, a behavior expected from an electrophilic intermediate.

C. Balancing Polar and Enthalpy Effects
The discussion at the beginning of this chapter focused on groups of similar reactions that obey the Evans-Polanyi relation, that is, reactions in which the energies of activation can be determined from reaction enthalpies using eq 1. Attention then turned to reactions where this simple relation (eq 1) does not hold. The energies of activation for reactions that do not obey the Evans-Polanyi relation are influenced by polar effects operative at the transition state. Since some reactions are more subject to enthalpy effects and others to polar effects, the question naturally arises as to what the balance is between these two. Principal component analysis answers this question with the finding that “the dominant factors influencing radical addition reactions are polar effects alone for strongly nucleophilic or strongly electrophilic radicals...and enthalpy effects alone for weakly nucleophilic or weakly electrophilic radicals”.18 For moderately nucleophilic or moderately electrophilic radicals both polar and enthalpy effects are important.18
.png?revision=1){kind=link}