
29.1: Molecular Orbitals of Conjugated Pi Systems
- Page ID
- 364595
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Frontier Molecular Orbital Theory
Prior to 1965, pericyclic reactions were known as "no mechanism reactions" since no one could adequately explain why reaction outcomes changed depending on whether reactants were exposed to heat or light. In 1965 Robert Burns Woodward and Roald Hoffmann used Frontier Molecular Orbital Theory, initially proposed by Kenichi Fukui, to develop their Theory of Conservation of Orbital Symmetry where outcomes of pericyclic reactions are explained by examining the Highest Occupied Molecular Orbital (HOMO) or Lowest Unoccupied Molecular Orbital (LUMO) of the reacting system. Their analysis of cycloadditions, electrocyclic reactions, and sigmatropic rearrangements is commonly referred to as the Woodward-Hoffmann Rules. A detailed analysis of three reaction types is provided in the subsequent sections of this chapter.
HOMO and LUMO are often referred to as frontier orbitals and their energy difference is termed the HOMO–LUMO gap. One common way of thinking about reactions in this way is through the concept of frontier orbitals. This idea says that if one species is going to donate electrons to another in order to form a new bond, then the donated electrons are most likely going to come from the highest occupied energy level. In this level, called the highest occupied molecular orbital (HOMO), the electrons are further from the nucleus and therefore less tightly held by the protons in the nucleus. The electrons would be donated, in turn, to the lowest empty energy level on the other species, called the lowest unoccupied molecular orbital (LUMO).
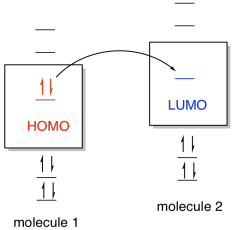
Molecular orbital interaction between frontier orbitals.
Molecular Orbitals
According to MO theory discussed in Section 1-11, when a double bond is non-conjugated, the two atomic 2pz orbitals combine to form two pi (π) molecular orbitals, one a low-energy π bonding orbital and one a high-energy π-star (π*) anti-bonding molecular orbital. These are sometimes denoted, in MO diagrams like the one below, with the Greek letter psi (Ψ) instead of π. In the bonding Ψ1 orbital, the two (+) lobes of the 2pz orbitals interact constructively with each other, as do the two (-) lobes. Therefore, there is increased electron density between the nuclei in the molecular orbital – this is why it is a bonding orbital. In the higher-energy anti-bonding Ψ2* orbital, the (+) lobes of one 2pz orbital interacts destructively with the (-) lobe of the second 2pz orbital, leading to a node between the two nuclei and overall repulsion. By the aufbau principle, the two electrons from the two atomic orbitals will be paired in the lower-energy Ψ1orbital when the molecule is in the ground state.

With a conjugated diene, such as 1,3-butadiene, the four 2p atomic orbitals combine to form four pi molecular orbitals of increasing energy. Two bonding pi orbitals and two antibonding pi* orbitals. The combination of four pi molecular orbitals allow for the formation of a bonding molecular orbital that is lower in energy than those created by an unconjugated alkene. The 4 pi electrons of 1,3-butadiene completely fill the bonding molecular orbitals giving is the additional stability associated with conjugated double bonds.
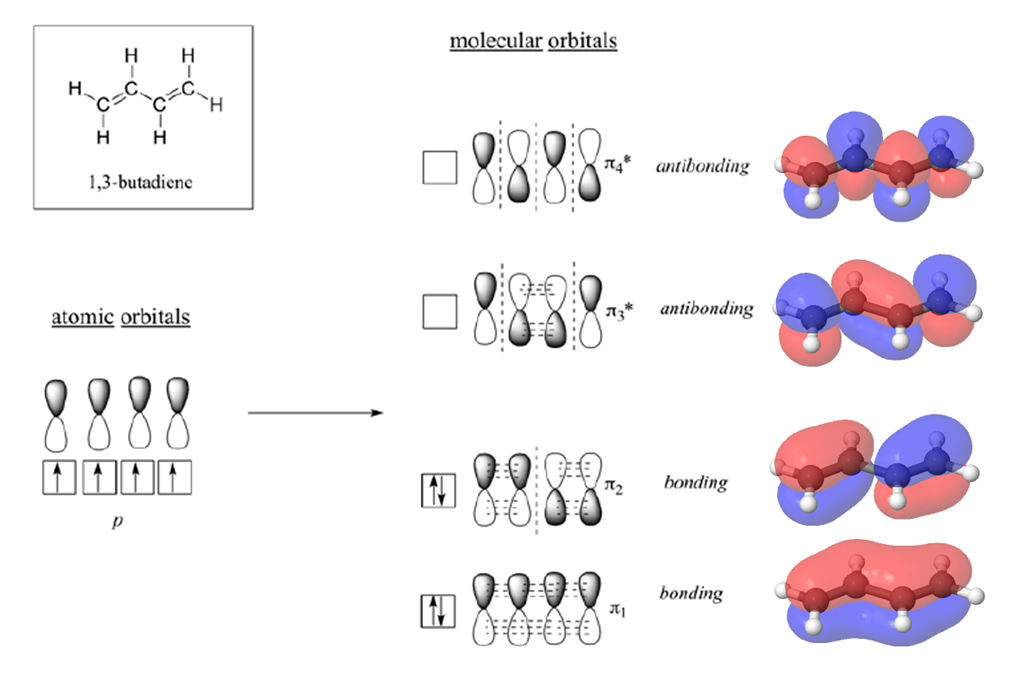
Electronic Transitions
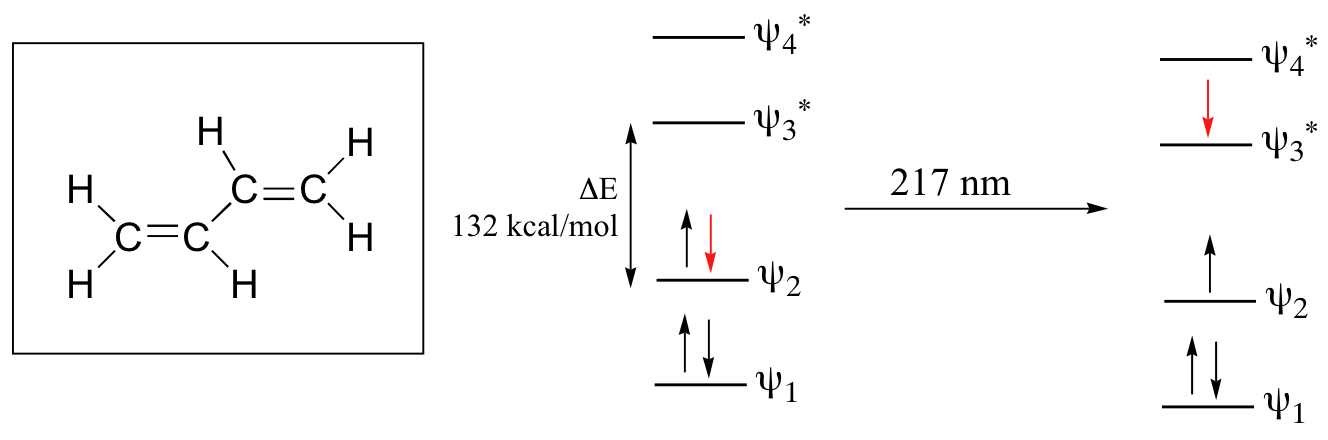
When a double-bonded molecule such as ethene (common name ethylene) absorbs 165 nm light, it undergoes a π - π* transition. An electron is moved from the HOMO of ethene to the LUMO placing the molecule in an excited state.

Where electronic transition becomes useful to most organic and biological chemists is in the study of molecules with conjugated pi systems. In these groups, the HOMO–LUMO gap energy gap for π -π* transitions is smaller than for isolated double bonds, and thus the wavelength absorbed is longer. The MO diagram for 1,3-butadiene, the simplest conjugated system. Recall that we can draw a diagram showing the four pi MO’s that result from combining the four 2pz atomic orbitals. The lower two orbitals are pi bonding, while the upper two are pi antibonding. Comparing this MO picture to that of ethene, our isolated pi-bond example the HOMO would be psi 2 and the LUMO would be psi 3. The HOMO-LUMO energy gap is smaller for the conjugated 1,3-butadiene system which absorbs UV light with a wavelength of 217 nm.
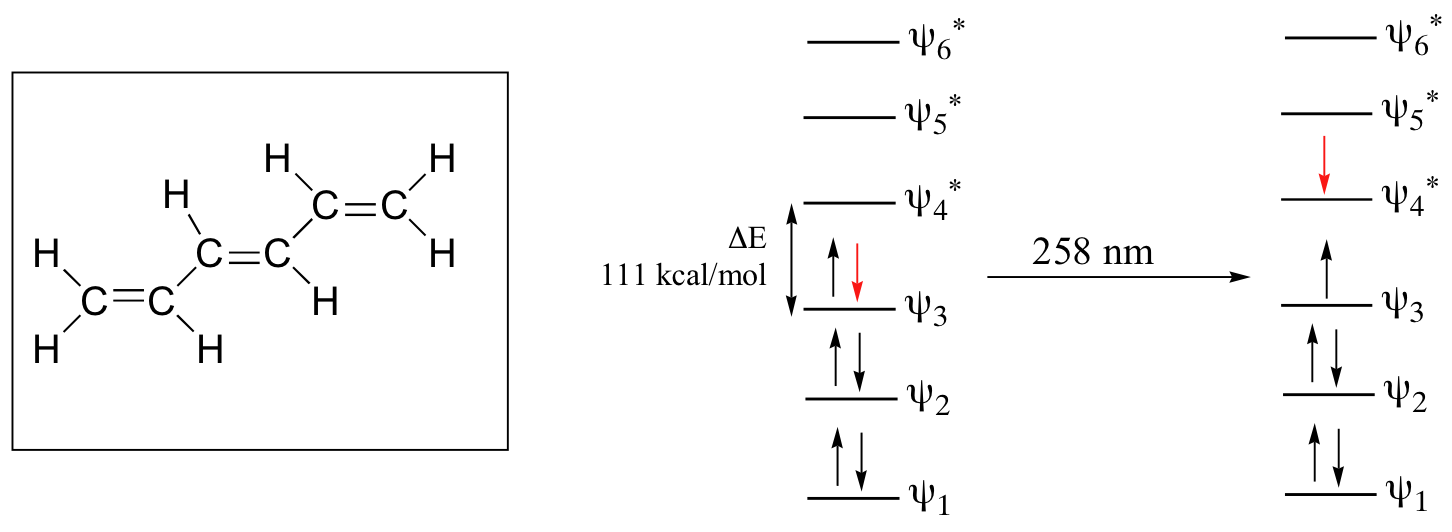
As conjugated pi systems become larger, the HOMO–LUMO gap energy gap for a π - π* transition becomes increasingly narrow, and the wavelength of light absorbed correspondingly becomes longer. The absorbance due to the π - π* transition in 1,3,5-hexatriene, for example, occurs at 258 nm.
Woodward-Hoffmann Rules
Much of what we have said about the electronic factors controlling pericyclic reaction was formulated in the mid 1960's by the American chemists R. B. Woodward and R. Hoffmann, in terms of what came to be called the orbital symmetry principles, or the Woodward-Hoffmann rules. This is a particularly simple approach says that many details of pericyclic reactions can explained by "conservation of orbital symmetry." This requires the symmetries of the molecular orbitals of reactants to be the same as the molecular orbitals of the products for a reaction to proceed.
The original approach of Woodward and Hoffmann involved construction of an "orbital correlation diagram" to see if the lobes of the reactant molecular orbitals match phases and allow for overlap required for bonding to occur. The symmetries of the appropriate reactant and product orbitals were matched to determine whether the transformation could proceed without a symmetry imposed conversion of bonding reactant orbitals to antibonding product orbitals. If the correlation diagram indicated that the reaction could occur without encountering such a symmetry-imposed barrier, it was termed symmetry allowed. If a symmetry barrier was present, the reaction was designated symmetry-forbidden.
Using the molecular orbital diagram for 1,3,5-hexatriene determine the HOMO and LUMO for both the ground and excited state.
- Answer
-
For the ground state the HOMO is psi 3 and LUMO is psi 4.
For the excited state the HOMO is psi 4 and LUMO is psi 5.