
15.7: Spectroscopy of Aromatic Compounds
- Page ID
- 31568
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)After completing this section, you should be able to
- determine whether an unknown compound contains an aromatic ring by inspection of its infrared spectrum, given a table of characteristic infrared absorptions.
- state the approximate chemical shift of aryl protons in a proton NMR spectrum.
- explain why signals resulting from the presence of aryl protons are found downfield from those caused by vinylic protons in a proton NMR spectrum.
- propose possible structures for an unknown aromatic compound, given its proton NMR spectrum, other spectroscopic data (such as a 13C NMR or infrared spectrum), or both.
Make certain that you can define, and use in context, the key term below.
- ring current
It is not necessary that you memorize detailed spectroscopic data. In the laboratory, on assignments and when writing examinations, you will be provided with a table of characteristic infrared absorptions to assist you in interpreting infrared spectra.
The important points to note about the proton NMR of aromatic compounds are the approximate chemical shifts of such protons and the complex splitting pattern that is sometimes observed. You are advised not to spend too long trying to understand why the signal for an aryl proton is found downfield from the signal for a vinylic proton. In general, we want you to be able to interpret NMR spectra, and leave the underlying theory for subsequent chemistry courses.
The chemical shifts of aromatic protons
Some protons resonate much further downfield than can be accounted for simply by the deshielding effect of nearby electronegative atoms. Vinylic protons (those directly bonded to an alkene carbon) and aromatic (benzylic) protons are dramatic examples.

We'll consider the aromatic proton first. Recall that in benzene and many other aromatic structures, a sextet of p electrons is delocalized around the ring. When the molecule is exposed to B0, these p electrons begin to circulate in a ring current, generating their own induced magnetic field that opposes B0. In this case, however, the induced field of the p electrons does not shield the benzylic protons from B0 as you might expect– rather, it causes the protons to experience a stronger magnetic field in the direction of B0 – in other words, it adds to B0 rather than subtracting from it.
To understand how this happens, we need to understand the concept of diamagnetic anisotropy (anisotropy means `non-uniformity`). So far, we have been picturing magnetic fields as being oriented in a uniform direction. This is only true over a small area. If we step back and take a wider view, however, we see that the lines of force in a magnetic field are actually anisotropic. They start in the 'north' direction, then loop around like a snake biting its own tail.
If we are outside the ring in the figure above, we feel a magnetic field pointing in a northerly direction. If we are inside the ring, however, we feel a field pointing to the south.
In the induced field generated by the aromatic ring current, the benzylic protons are outside the ring – this means that the induced current in this region of space is oriented in the same direction as B0.
In total, the benzylic protons are subjected to three magnetic fields: the applied field (B0) and the induced field from the electrons pointing in one direction, and the induced field of the non-aromatic electrons pointing in the opposite (shielding) direction. The end result is that benzylic protons, due to the anisotropy of the induced field generated by the ring current, appear to be highly de-shielded. Their chemical shift is far downfield, in the 6.5-8 ppm region.
The presence of anisotropy effects is often used to indicate aromaticity in a molecule. An example is the nonaromatic 8 pi electron cyclooctatetraene. In order to avoid anti-aromatic destabilization, the molecule takes a non-planar conformation which prevents conjugation and ring currents. Consequently, the molecule's protons absorbed at 5.8 ppm which is within the alkene region of 1H NMR.
The 1H-NMR spectrum of [18] annulene has two peaks, at 8.9 ppm and -1.8 ppm (upfield of TMS!) with an integration ratio of 2:1. Explain the unusual chemical shift of the latter peak.
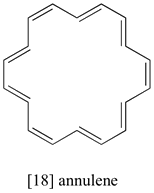
- Answer
-
The molecule contains two groups of equivalent protons: the twelve pointing to the outside of the ring, and the six pointing into the center of the ring. The molecule is aromatic, as evidenced by the chemical shift of the ‘outside’ protons’ (and the fact that there are 18 p electrons - a Hückel number). The inside protons are shielded by the induced field of the aromatic ring current, because inside the ring this field points in the opposite direction of B0. This is the source of the unusually low (negative relative to TMS) chemical shift for these protons.
Characteristic 1H NMR Absorptions of Aromatic Compounds
Protons directly attached to an aromatic ring, commonly called aryl protons, show up about 6.5-8.0 PPM. This range is typically called the aromatic region of an 1H NMR spectrum. Protons on carbons directly bonded to an aromatic ring, called benzylic protons, show up about 2.0-3.0 PPM.
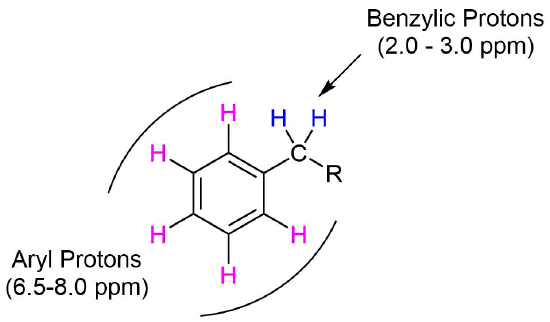 '
'
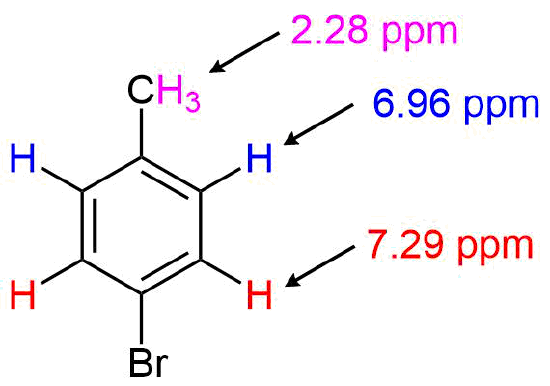
For the 1H NMR spectrum of p-bromotoluene, the absorption for the benzylic protons appears as a strong singlet at 2.28 ppm. Due to the molecule's symmetry, the aryl protons appear as two doublets at 6.96 & 7.29 ppm.

Characteristic 13C NMR Absorptions of Aromatic Compounds
Carbons in an aromatic ring absorb in the range of 120-150 ppm in a 13C NMR spectrum. This is virtually the same range as nonaromatic alkenes (110-150 ppm) so peaks in this region are not definitive proof of a molecule's aromaticity.
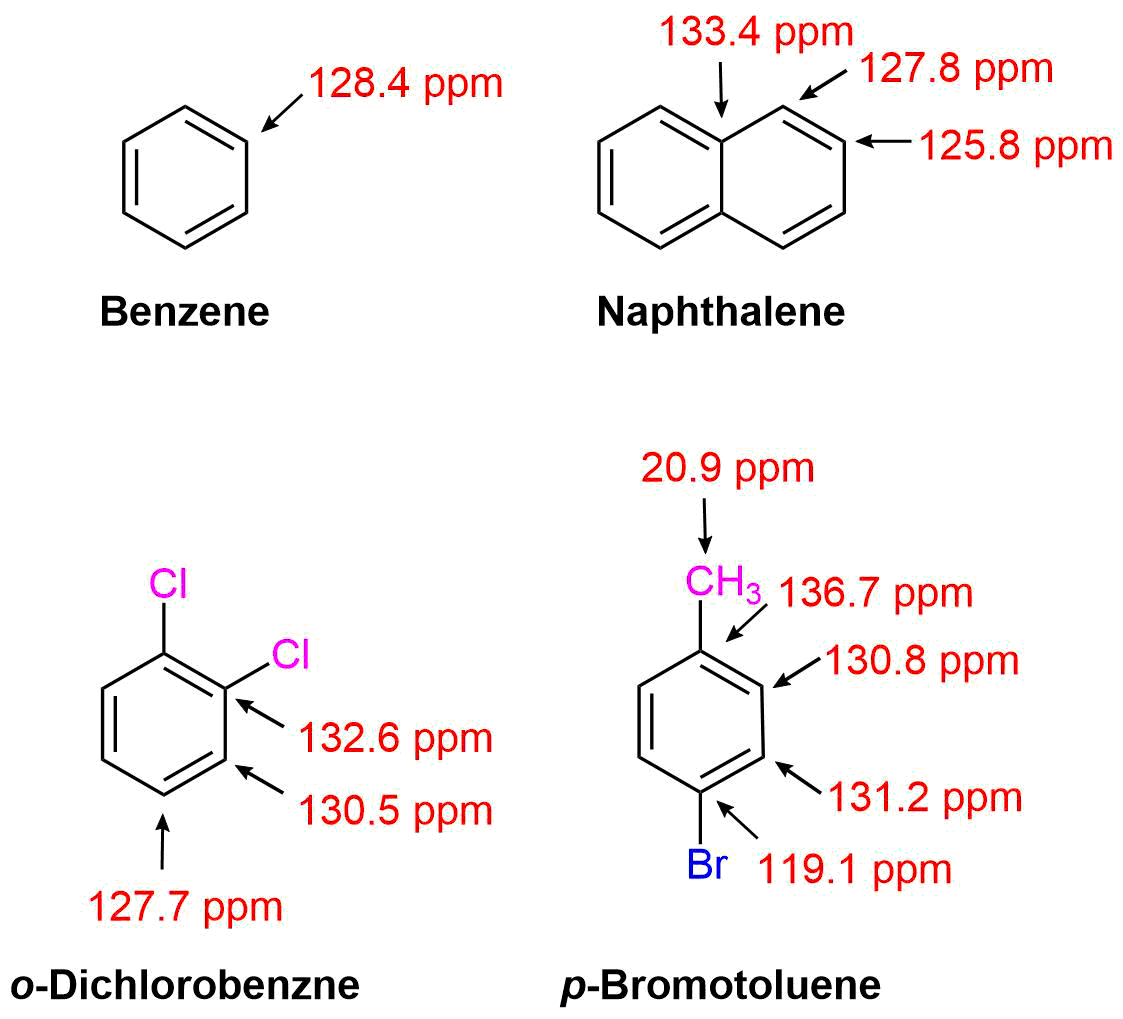
Due to the decoupling in 13C NMR, the number of absorptions due to aromatic carbons can easily be observed. This can be used to determine the relative positions (ortho, meta, or para) for di-substituted benzenes. Peak assignments can be simplified by noting that 13C peaks tend to be larger if two carbons contribute to the absorption.
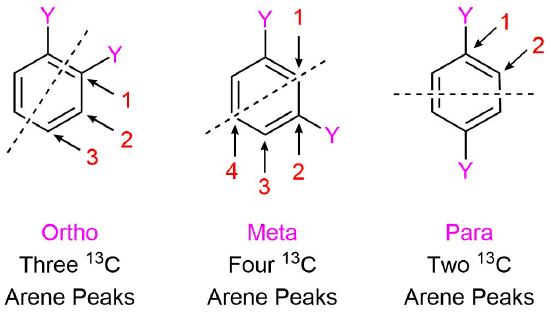
Symmetrical Di-substituted Benzenes
Benzenes substituted with two identical groups have a relatively high amount of symmetry. In all three configurations, there is a plane of symmetry which reduces the number of distinct aryl carbon absorptions to less than six. The ortho configuration has a plane of symmetry which mirrors each carbon in the benzene ring causing only three 13C aryl absorptions to occur. The meta configuration's plane of symmetry mirrors two carbons of the benzene ring allowing for four aryl absorptions to occur. The para configuration actually has two planes of symmetry (one vertical and one horizontal on the structure below) through the benzene ring, which only allows two distinct aryl absorptions to occur.
Asymmetrical Di-substituted Benzenes
The lack of a plane of symmetry in asymmetrical di-substituted benzenes makes each carbon in the ortho and meta configuration unique. Consequently, their 13C NMR spectra show six arene absorptions. However, the para configuration has a plane of symmetry drawn through the two substituents which mirrors two carbons of the benzene ring causing only 4 arene absorptions to appear.
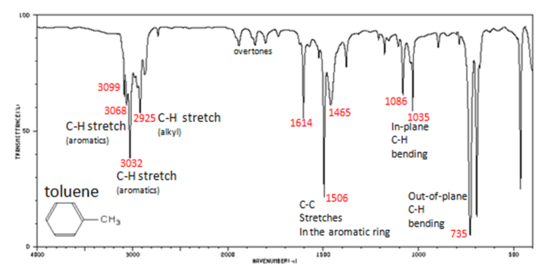
Charateristic IR Absorption of Benzene Derivatives
Arenes have absorption bands in the 650-900 cm−1 region due to bending of the C–H bond out of the plane of the ring. The exact placement of these absorptions can indicate the pattern of substitution on a benzene ring. However, this is beyond the scope of introductory organic chemistry. Arenes also possess a characteristic absorption at about 3030-3100 cm−1 as a result of the aromatic C–H stretch. It is somewhat higher than the alkyl C–H stretch (2850–2960 cm−1), but falls in the same region as olefinic compounds. Two bands (1500 and 1660 cm−1) caused by C=C in plane vibrations are the most useful for characterization as they are intense and are likely observed.
In aromatic compounds, each band in the spectrum can be assigned:
- C–H stretch from 3100-3000 cm-1
- overtones, weak, from 2000-1665 cm-1
- C–C stretch (in-ring) from 1600-1585 cm-1
- C–C stretch (in-ring) from 1500-1400 cm-1
- C–H "oop" from 900-675 cm-1
Note that this is at slightly higher frequency than is the –C–H stretch in alkanes. This is a very useful tool for interpreting IR spectra. Only alkenes and aromatics show a C–H stretch slightly higher than 3000 cm-1.
Ultraviolet Spectroscopy
The presence of conjugated π bonds makes aromatic rings detectable in a UV/Vis spectrum. Benzene primarily absorbs through a be a π-π* transition over the range from 160-208 nm with a λ max value of about 178 nm.
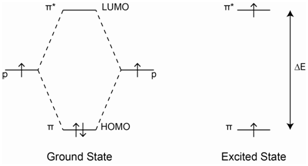
Benzene shows a less intense absorption in 230-276 nm range. These absorptions are due to vibrational fine structure often displayed by rigid molecules such as benzene and other aromatic compounds.