
9.3: Reactions of Alkynes - Addition of HX and X₂
- Page ID
- 31483
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)After completing this section, you should be able to
- describe the bonding and geometry of the carbon-carbon triple bond in terms of the sp-hybridization of the carbon atoms involved.
- explain the reactivity of alkynes based on the known strengths of carbon-carbon single, double and triple bonds.
- write equations for the reaction of an alkyne with one or two equivalents of halogen (chlorine or bromine) or halogen acid (HCl, HBr or HI).
- draw the structure of the product formed when an alkyne reacts with one equivalent of the halogens and halogen acids listed in Objective 3.
- identify the alkyne which must have been used in an addition reaction with a halogen or halogen acid, given the product of such a reaction.
You might find it useful to review Section 1.9 before you begin work on this chapter. If necessary, construct a molecular model of a simple alkyne. Notice the similarity between the behaviour of alkenes and that of alkynes. In the laboratory, you will observe that alkynes readily decolourize a solution of bromine in dichloromethane. Section 9.7 describes a test that allows you to distinguish between a terminal alkyne (i.e., one in which the triple bond occurs between the last two carbons in the chain) and nonterminal alkynes and alkenes.
The Alkyne Triple Bond
As discussed in Section 1-9, the carbon-carbon triple bonds of alkynes are created by the the overlap of orbitals on two sp hybridized carbon atoms. The molecule acetylene (HCCH) is said to contain three sigma bonds and two pi bonds. The C-C sigma bond of acetylene is formed by the overlap an sp hybrid orbital from each of the carbon atoms. The two C-H sigma bonds are formed by the overlap of the second sp orbital on each carbon atom with a 1s orbital from a hydrogen. Each carbon atom still has two half-filled P orbitals, which are perpendicular both to each other and to the line formed by the sigma bonds. These two perpendicular pairs of p orbitals form two pi bonds between the carbons, resulting in a triple bond overall (one sigma bond plus two pi bonds). The electrostatic potential map of acetylene shows that the pi electrons of the triple bond form a negative belt (shown in red) around the center of the molecule.

Acetylene is linear, as predicted by VSEPR, with all four atoms lying in a straight line and both H-C-C bond angles being 180o. The triple bond in acetylene is the shortest (120 pm) and the strongest (964 kJ/mol) carbon-carbon bond known.

Electrophilic Addition of HX to Alkynes
Alkynes undergo electrophilic addition in much the same manner as alkenes, however, the presence to two pi bonds allows for the possibly of the addition happening twice. The addition of one equivalent of hydrogen chloride or hydrogen bromide converts alkynes to haloalkenes. The addition of two or more equivalents of HCl or HBr converts alkynes to geminal dihalides through an haloalkene intermediate. These additions are regioselective and follow Markovnikov's rule. The double bonds formed during the reaction with internal alkynes tend to have Z stereochemistry although not always.
HBr Addition to a Terminal Alkyne

HCl Addition to a Symmetrical Internal Alkyne
-4-chlorohex-3-ene%253B_hex-3-yne_reacts_with_2_equivalents_of_HCl_in_acetic_acid_to_give_3%252C3-dichlorohexane.svg?revision=3&size=bestfit&width=611&height=254)
HBr Addition to an Asymmetrical Internal Alkyne
The addition of HX to an asymmetrical internal alkyne tend to make a mixture of isomers as products.
-3-bromopent-2-ene_and_(Z)-2-bromopent-2-ene%253B_pent-2-yne_reacts_with_2_equivalents_of_HBr_in_acetic_acid_to_give_3%252C3-dibromopentane_and_2%252C2-dibromopentane.svg?revision=3&size=bestfit&width=795&height=253)
Mechanism
The mechanism for the electrophilic addition of HX to an alkyne is analogous to the HX addition to an alkene. The presence of two pi bonds in the alkyne allows for the addition of HX to occur twice. The addition of H+ to the alkyne forms a vinyl cation will preferably form on the more substituted side of the alkyne following Markovnikov's rule. The subsequent addition of Br- forms a haloalkene which undergoes electrophilic addition to a second H+. The carbocation form will preferably form on the carbon attached to the halogen already in place. The carbocation is stabilized by the halogen through the creation of a resonance structure which obeys the octet rule. This stabilizing effect ensures that a geminal-dihalide is the sole product and no vicinal-dihalide is formed.

Electrophilic Addition of X2 to Alkynes
Alkynes undergo the same type of electrophilic addition with chloride and bromine as alkenes. However, with alkynes the halogen addition can occur once or twice depending on the molar equivalents of halogen used in the reaction. If one molar equivalent of halogen is used, a dihaloalkene is formed. The anti addition of the reaction mechanism causes the halogens to be trans in the resulting alkene. The addition of two or more molar equivalents of halogen converts the alkyne to a tetrahaloalkane through a dihaloalkene intermediate.
-1%252C2-dibromopent-1-ene%252C_and_pent-1-yne_reacts_with_2_equiv_of_Br2_in_dichloromethane_to_give_1%252C1%252C2%252C2-tetrabromopentane.svg?revision=2&size=bestfit&width=525&height=249)
Mechanism
The alkyne undergoes electrophilic addition with bromine to form a bromonium ion in a three-membered ring. The ejected bromide ion performs an SN2 reaction with the bromonium ion causing the ring to open and the bromines in the resulting alkene to be in a trans configuration. The process is repeated with a second pi bond creating a tetrahaloalkane as a product.

Relative Reactivity of Alkynes and Alkenes to Electrophilic Reagents
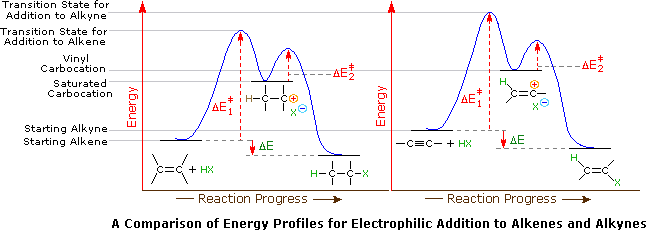
When the addition reactions of electrophilic reagents, such as strong Brønsted acids and halogens, to alkynes are studied there is a curious paradox. The reactions of alkynes are even more exothermic than the additions to alkenes, and yet the rate of addition to alkynes is slower by a factor of 100 to 1000. This concept is shown in the reaction of one equivalent of bromine with 1-penten-4-yne to produce 4,5-dibromopent-1-yne as the chief product.

Why are the reactions of alkynes with electrophilic reagents more sluggish than the corresponding reactions of alkenes? Typically, addition reactions to alkynes are more exothermic than additions to alkenes, and there would seem to be a higher π-electron density about the triple bond ( two π-bonds versus one ). Two factors are significant in explaining this apparent paradox. First, although there are more π-electrons associated with the triple bond, the sp-hybridized alkyne carbons are more electronegative than the sp2-hybridized alkene carbons. The alkyne carbons exert a strong attraction for their π-electrons, which are consequently bound more tightly to the functional group than are the π-electrons of a double bond. This is seen in the ionization potentials of ethylene and acetylene. Remember an ionization potential is the minimum energy required to remove an electron from a molecule of a compound. Since the initial interaction between an electrophile and an alkene or alkyne involves the donation of electrons, the relatively slower reactions of alkynes becomes understandable.
| Acetylene | HC≡CH + Energy → [HC≡CH •(+) + e(–) | ΔH = +264 kcal/mole |
|---|---|---|
| Ethylene | H2C=CH2 + Energy → [H2C=CH2] •(+) + e(–) | ΔH = +244 kcal/mole |
A second factor is presumed to be the stability of the carbocation intermediate generated by sigma-bonding of a proton or other electrophile to one of the triple bond carbon atoms. When comparing the mechanism for the addition of HBr to an alkene and an alkyne, the alkyne reaction creates a vinyl carbocation which is less stable than the alkyl carbocation made during the alkene reaction.

Indeed, we can modify our earlier ordering of carbocation stability to include these vinyl cations in the manner shown below.
| Substitution | Methyl | 1°-Vinyl | 1° | 2°-Vinyl | 2° | 1°-Allyl | 3° | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Stability | CH3(+) | ≈ | RCH=CH(+) | < | RCH2(+) | ≈ | RCH=CR(+) | < | R2CH(+) | ≈ | CH2=CH-CH2(+) | < | R3C(+) |
Application of the Hammond postulate indicates that the activation energy for the generation of a vinyl cation intermediate would be higher than that for a lower energy intermediate. Thus, electrophilic reactions with alkenes have a lower activation energy and process faster than the corresponding reaction with an alkyne. This is illustrated for alkenes versus alkynes by the following energy diagrams. Despite these differences, electrophilic additions to alkynes have emerged as exceptionally useful synthetic transforms.
Electrophilic Addition of HX with Peroxides to Alkynes
When 1 equivalent of HBr is reacted with alkynes in the presence of peroxides and Anti-Markovnikov addition occurs. The use of peroxides causes the reaction to occur via a free radical mechanism. The bromine adds to the less substituted alkyne carbon while the hydrogen adds to the more substituted creating a haloalkene. Typically, H and Br are added in a syn or anti manner creating a mixture of products.
_and_(E)-1-bromopent-1-ene.svg?revision=1&size=bestfit&width=761&height=105)
Draw the structure and give the IUPAC name of the product formed in each of the reactions listed below:

- Answer
-
