
Sonogashira Coupling
- Page ID
- 69102
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
The Sonogashira reaction (also called the Sonogashira-Hagihara reaction) is the cross coupling of aryl or vinyl halides with terminal alkynes to generate conjugated enynes and arylalkynes (Scheme 1). The reaction typically proceeds in the presence of a palladium(0) catalyst, a copper(I) cocatalyst, and an imine base.1 Alternative procedures describe Sonogashira coupling reactions performed without the Cu(I) cocatalyst.
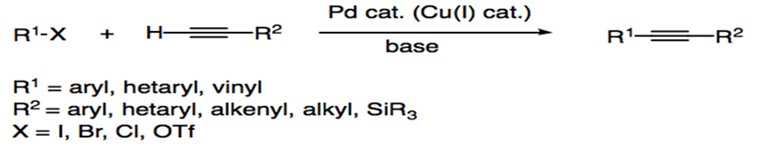
Scheme 1: General reaction for Sonogashira Coupling.
Preceding the Sonogashira reaction, Heck and Diek and Cassar reactions reported the arylation or alkenylation of alkenes via phosphane-palladium complexes.2, 3 However, both procedures required harsh reaction conditions, such as high temperatures (100oC). In 1975, Kenkichi Sonogashira, Yasuo Tohda, and Nobue Hagihara reported the cross coupling of iodobenzene and acetylene in the presence of Pd(Ph3)4Cl2 and CuI.4 The use of the Cu+ cocatalyst, a procedural design that built on the work of Castro and Stephens,5 increased the reactivity of the reagents and allowed the reaction to proceed at room temperature. As a result, the Sonogashira reaction remains one of the most popular cross-coupling reactions for the formation of C(sp2)-C(sp) bonds, with its use extending to applications in pharmaceuticals, nanomaterials, and natural products. In fact, Chinchilla and Najera (2007) reported a significant increase in interest for Sonogashira coupling within the past thirteen years.1 Efforts within the field aim to further improve reaction conditions, catalyst designs, and mechanistic understandings of Sonogashira coupling.
MECHANISM
Palladium and Copper Co-catalyzed Cycle
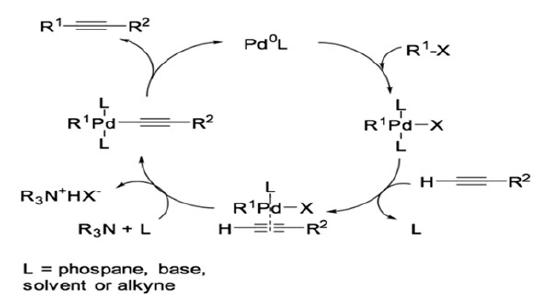
Palladium and copper co-catalyzed Sonogashira coupling is reported to proceed via two independent catalytic cycles (Scheme 2).6-18

Scheme 2: Catalytic cycle of palladium and copper co-catalyzed Sonogashira reaction.
Prior to oxidative addition, the 14-electron PdL2 complex is generated through a reductive process known as σ-complexation-dehydropalladation-reductive elimination. In this process, palladium(II) is reduced by forming a complex with electron donor compounds that function as either ligands or solvent within the reaction (Scheme 3).
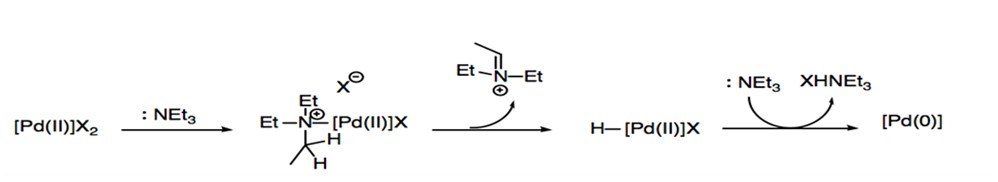
Scheme 3: σ-complexation-dehydropalladation-reductive elimination with amide.
The Pd0 complex then undergoes oxidative addition of R1-X to form a four-coordinated palladium complex. At this point in the reaction, the palladium cycle intersects with the copper cycle. For the co-catalyzed copper cycle, the base typically used is amide. Due to the relatively low basicity of the amide, a π-alkyne-copper complex must be formed to increase the acidity of the alkyne for the alkyne to undergo deprotonation. Following deprotonation in the copper-catalyzed cycle, copper acetylide is formed. It is noteworthy that the formation of the palladium acetylide from the copper acetylide and four-coordinated palladium complex is typically considered the rate determining step. This step is called transmetalation. Trans/cis isomerization occurs next, which is then followed by reductive elimination to afford the final product.1
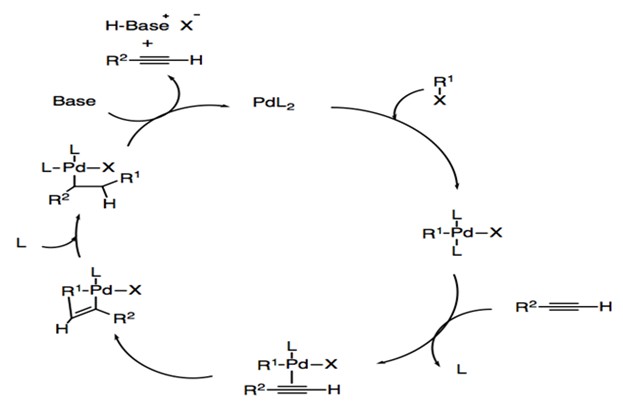
Copper-free Catalytic Cycle
For copper-free Sonogashira coupling (Scheme 4), the first step is also the oxidative addition to form the four-coordinated palladium complex. Because the basicity of amine is insufficient for the deprotonation of alkyne, the dissociation of a neutral ligand and the formation of a π-alkyne-palladium complex occurs.19 This is followed by deprotonation of the alkyne to form palladium acetylide. Then trans/cis isomerization and reductive elimination occur stepwise to generate the final product and Pd0L2 catalyst.
Scheme 4: Catalytic cycle of copper-free Sonogashira reaction.
Mechanistic Studies
While the exact mechanism of Sonogashira coupling is not known, the catalytic cycles depicted in Scheme 2 are generally accepted. Efforts to isolate and characterize proposed palladium intermediates of the homogeneous Sonogashira reaction have proven difficult, yet some transient species have successfully been identified with multinuclear NMR spectroscopy.1 Alternative methods have turned towards using heterogeneous catalysts and analyzing surface transient organometallic intermediates, as well as performing kinetic measurements using gas chromatography, to corroborate the proposed mechanism.20-22
The real catalyst involved in the catalytic cycle is still under debate. A monoligated palladium catalyst can be observed when the neutral ligand is bulky, suggesting that the catalyst undergoes dissociation prior to oxidative addition.23 It has also been shown that the Pd0L2 will form a corresponding anionic palladium complex if the solution contains anions such as halides. For instance, [L2Pd0Cl]- can be generated from Pd0L2 in the presence of a chloride ion.24 Therefore, it is possible that an anionic palladium complex is the real catalyst in the catalytic cycle.25
As discussed previously, amide is typically the base used in the Sonogashira reaction, and it is proposed that a π-alkyne-copper complex is formed to increase the acidity of the alkyne. However, this proposed π-alkyne-copper complex has not been directly observed.24 In 2005, Berger et al observed a π-alkyne-silver complex using NMR in the palladium and silver co-catalyzed Sonogashira coupling.25 Experimental reports such as this provide evidence for a similar π-alkyne complex forming in the palladium and copper co-catalyzed coupling reaction.
The mechanism for copper-free Sonogashira coupling has been studied with chemical kinetic experiments and density functional theory (DFT) calculations. In particular, DFT calculations have recently proposed an alternative mechanistic pathway, named carbopalladation (Scheme 5). However, the DFT calculations and theoretical mechanistic studies concluded that the activation barrier for this cycle is too high, and subsequently supported Scheme 3 as the observed mechanism.26
Scheme 5: Carbopalladation cycle for copper-free Sonogashira coupling.
Regio- and Stereoselectivity
Disubstituted aryl or vinyl halides have allowed studies to probe at the regioselectivity of Sonogashira coupling. For compounds with two different halides, such as 2-bromo-4-iodo-quinoline, the acetylene adds to the site with the more reactive iodide substituent (Scheme 6). Substrates that have the same halide substituent demonstrate alkynylation at the more electrophilic site (Scheme 7).27
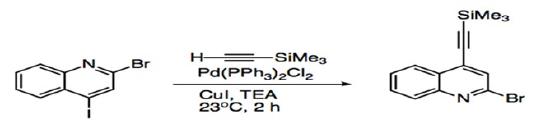
Scheme 6: Sonogashira coupling of disubstituted quinoline.
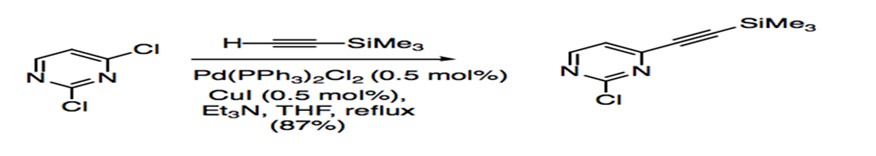
Scheme 7: Sonogashira coupling of dichloro aryl substrate.
Sonogashira coupling offers a straightforward synthetic route with regards to stereochemistry. The formation of the C(sp2)-C(sp) bonds does not generate new stereocenters in the final product, and no migrations have been reported.28 Additionally, the stereochemical information present in the starting materials is retained in the final product (Scheme 8).29
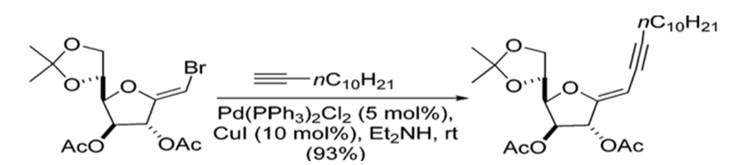
Scheme 8: Preparation of enyne where stereochemistry is retained.
REACTION CONDITIONS
Substrate Scope
The first step of the catalytic cycle is oxidative addition of the aryl or vinyl halide to the phosphane-palladium complex. The general trend of substrate reactivity towards oxidative addition follows:
vinyl iodide ≥ vinyl triflate > vinyl bromide > vinyl chloride > aryl iodide > aryl triflate ≥ aryl bromide >> aryl chloride
Activation of aryl chlorides is particularly difficult due to the lower electrophilicity of the compound. Despite the favorable reactivity of iodo-substrates, these reagents are expensive and unstable compared to the other halide and triflate compounds. Less reactive substrates have been modified to increase activation towards oxidative addition. Such modifications include placing electron withdrawing groups (EWGs) on R1 to decrease electron density between R1 and X (Scheme 1). The reactivity towards oxidative addition also proceeds more favorably with the use of electron rich phosphane ligands.
The terminal alkyne substrate exhibits a relatively broad range of functional group compatibility. Sonogashira et al originally used acetylene, but subsequent studies have shown that the alkyne can accommodate aryl, hetaryl, alkenyl, alkyl, and trialkylsilyl functional groups. In some cases, the functional group of the alkyne helps facilitate Sonogashira coupling in the absence of the copper cocatalyst. For example, trimethylsilylacetylene has been used to generate the terminal acetylene in a process known as “sila”-Sonogashira coupling.30
Ligand Design and Applications
Changes in catalytic activity and stability of palladium catalysts in Sonogashira reactions can be achieved by modifying the catalyst ligands. Electron-rich phophane ligands, instead of the commonly used triphenylphosphane ligands, can increase the rate of oxidative addition of the aryl halides.1 Additionally, sterically bulky ligands can promote the dissociation of the active palladium catalyst from the resting state.31 Thus, bulky ligands are used to generate corresponding palladium complexes from weakly coordinated palladium complexes, such as Pd(OAc)2, Pd2(dba)3. The combination of bulky and electron rich ligands achieves more efficient Sonogashira couplings. Ligand 1, both bulky and electron rich, is used in the copper-free Sonogashira reaction in the presence of (PhCN)2Cl2 and cesium carbonate as a base. 32 Ligand 2 can catalyze the co-catalyzed Sonogashira reaction of aryl iodides at room temperature when combined with Pd2(dba)3.33 Furthermore, the corresponding palladium catalyst generated from Pd(OAc)2 and bis-(tert-butyl)aminomethylphosphane, ligand 3, is reactive enough that it can catalyze the copper-free Sonogashira reaction of aryl chloride efficiently.34
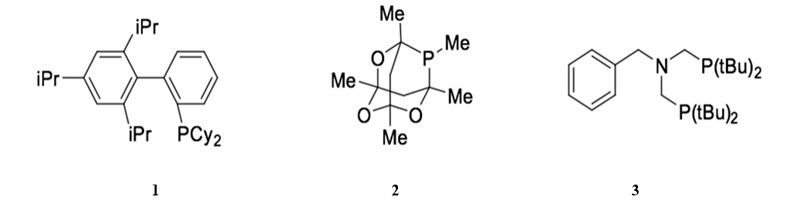
Classification and properties of palladium catalysts
Palladium-phosphorus complexes.
Typically, Sonogashira reactions proceed under homogeneous reaction conditions in the presence of palladium phosphorus complexes and copper(I) catalysts. The most common catalyst used in Sonogashira reactions is Pd(PPh3)4 and Pd(PPh3)2Cl2, although bidentate ligands have also been used, such as Pd(dppe)Cl2, Pd(dppp)Cl2, or Pd(dppf)Cl2. While Pd(PPh3)2Cl2 is more soluble and stable than Pd(PPh3)4, both catalysts require up to 5% loading to afford good yield.1 Therefore, an important aspect of the field is developing more efficient palladium phosphorus catalysts. For instance, it has been shown that the mixing of Pd2(dba)3/P(t-Bu)3 can catalyze the cross-coupling of terminal alkyne and p-toluenesulfonyl chloride to generate the corresponding alkyne in good yield in the presence of potassium carbonate (Scheme 9).35
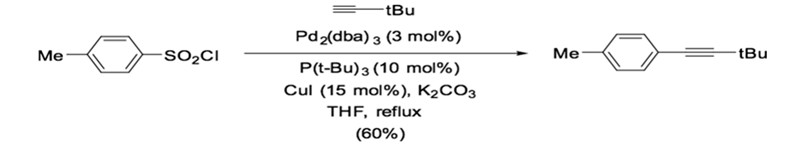
Scheme 9: Palladium phosphorus complex catalyzed desulfitative cross-coupling.
Palladium-nitrogen complexes
Palladium-nitrogen catalysts are also effective for Sonogashira coupling, particularly for copper-free reactions.36 Catalyst 3, a bis-imidazolyl-derived palladium complex, can facilitate the coupling of terminal alkynes and aryl iodides with very low catalyst loading (0.02 mol %) when used in the presence of piperidine.37 Similarly, bis-oxazoline palladium catalyst 4 is used for the cross coupling of phenylacetylene and iodobenzene with low catalyst yield (0.055 mol %) in the presence of CuI.38, 39 The dipyrimidyl-palladium catalyst 5 can be used in the cross coupling of aryl diiodides or aryl dibromide to achieve double coupling with phenylacetylene in good yield (Scheme 10). 40
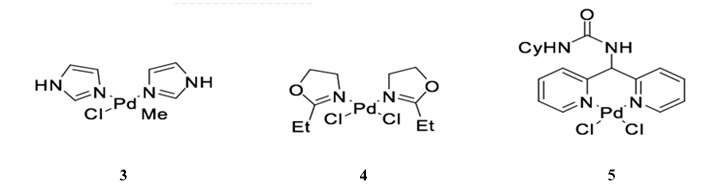
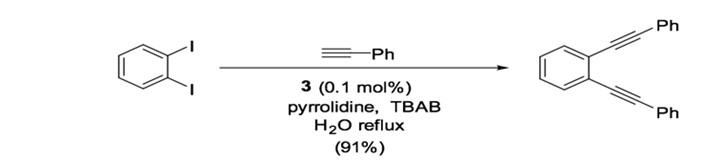
Scheme 10: Dipyrimidyl-palladium complex catalyzed Sonogashira reaction.
N-Heterocyclic Carbene (NHC) Palladium Complexes.
N-Heterocyclic carbenes (NHCs) can efficiently replace phosphane ligands on palladium complexes due to their ability to behave similarly to σ-donor ligands. Catalyst 6 can catalyze the Sonogashira reaction of aryl bromides in the presence of PPH3 and CuI.41 Bis-carbene palladium catalyst 7 has also been reported to catalyze copper co-catalyzed the Sonogashira coupling of aryl bromides with alkynes in boiling pyrrolidine with good yield.42
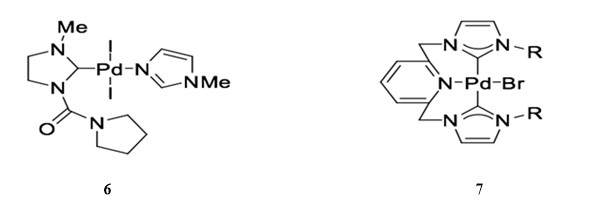
NHC palladium catalysts can also be used to catalyze copper-free Sonogashira reactions. For example, NHC-derived catalyst 8 is used for the cross-coupling of phenylacetylene and 2-bromoacetopheone in the presence of triethylamine.43 NHC-derived palladacycle 9 is used for the same copper-free Sonogashira reaction with relatively low catalyst loading (0.1 mol %), although the reaction achieves relatively low yield.44 In general, though, palladacycle complexes are stable and demonstrate favorable catalytic activity in coupling reactions.

LIMITATIONS
Despite the popularity of the reaction, Sonogashira coupling exhibits several important limitations. The copper salts used in the co-catalyzed reaction are environmentally unfriendly and difficult to recover from the reaction mixture. Additionally, upon exposure to air, the copper acetylide will undergo a homocoupling side reaction, decreasing the overall efficiency of the reaction. 45
Although copper-free Sonogashira reactions can avoid some problems discussed, this kind of reaction is also environmentally unfriendly because it requires the high loading of bases such as amines, 1 and it is also reported that some commercially available palladium catalysts such as PdCl2 contain trace amount of copper salt. Therefore, it is possible that copper-free Sonogashira reactions are not truly “copper-free,” and a trace amount of copper salt may still have a notable influence on the overall reaction. 46
APPLICATIONS IN SYNTHESIS
The mild reaction conditions and relatively broad range of substrate compatibility allow Sonogashira coupling to find use in a great number of synthetic applications. As a reaction that retains the stereospecific information contained in the starting material, Sonogashira coupling is particularly useful for the generation of enynes and enediynes, an important component for natural products. Many metabolites require the incorporation of enyne moieties. The enyne moiety is generated by coupling a vinylic halide with an alkyne. Sonogashira coupling is used in the synthesis of (-)-isoprelaurefucin, a metabolite that comes from red algae. The intermediate of this total synthesis is achieved with the coupling of an vinyl iodide with trimethylsilylacetylene (Scheme 11).1 The total syntheses of natural products such as benzylisoquinolines, indole alkaloids, and benzofuropyranones also use Sonogashira coupling to form intermediate products. For example, the Sonogashira protocol is used for the coupling of aryl iodide to an aryl acetylene to eventually form bulgaramine, a benzindenoazepine alkaloid (Scheme 12).1
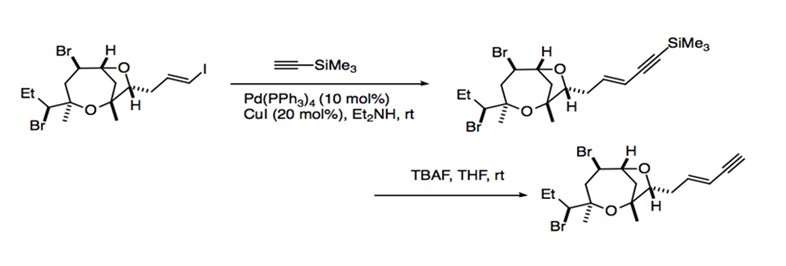
Scheme 11: Synthesis of (-)-isoprelaurefucin.
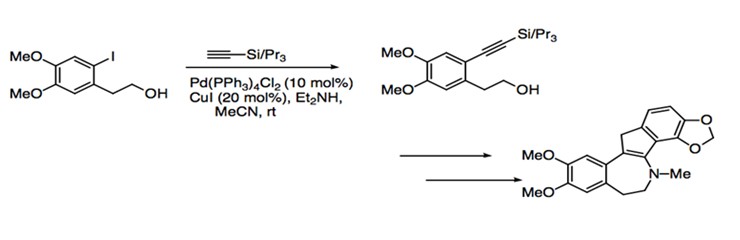
Scheme 12: Synthesis of bulgaramine.
While an important reaction in the synthesis of natural products, Sonogashira coupling also plays an important role in materials chemistry. The alkyne substrates provides the opportunity to generate extended networks of highly conjugated pi systems, giving rise to eletrooptical and electronic properties in molecules. Poly(phenyleneethynylene)s (PPEs) are synthesized in a one-pot polymerization via the coupling of aryl alkynes with aryl iodides (Scheme 13).1
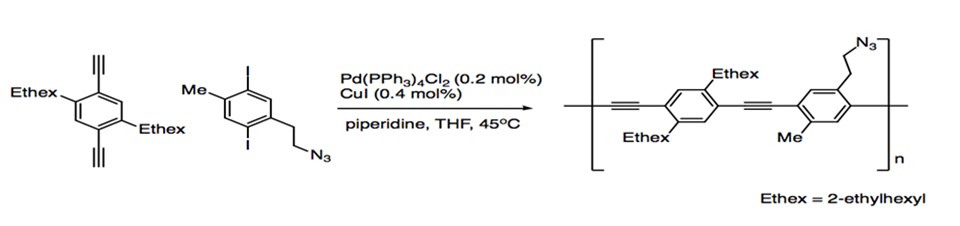
Scheme 13: Polymerization with Sonogashira Coupling.
REFERENCES
- Chinchilla, R.; Najera, C. Chem. Rev. 2007, 107, 874.
- Diek, H. A.; Heck, F. R. J. Organomet. Chem. 1975, 93, 259.
- Cassar, L. J. Organomet. Chem. 1975, 93, 253.
- Sonogashira, K.; Tohda, Y.; Hagihara, N. Tetrahedron Lett. 1975, 16, 4467.
- Stephens, R. D.; Castro, C. E. J. Org. Chem. 1963, 28, 2163.
- Zapf., A.; Beller, M. Top. Catal. 2002, 19, 101.
- Tucker, C. E.; de Vries, J. G. Top. Catal. 2002, 19, 111.
- Brase, S.; Kirchhoff, J. H.; Kobberling, J. Tetrahedron 2003, 59, 885.
- van de Weghe, P. Lett. Org. Chem. 2005, 2, 113.
- Brandsma, L. Synthesis of Acetylenes, Allenes and Cumulenes: Methods and Techniques; Elsevier: Oxford, 2004; p 293.
- Sonogashira, K. In Metal-Catalyzed Cross-Coupling Reactions; Diederich, F., de Meijera, A., Eds.; Wiley-VCH: Weinheim, 2004; Vol. 1, p 319.
- Tykwinski, R. R. Angew. Chem., Int. Ed. 2003, 42, 1566.
- Negishi, E.; Anastasia, L. Chem. Rev. 2003, 103, 1979.
- Sonogashira, K. In Handbook of Organopalladium Chemistry for Organic Synthesis; Negishi, E., de Meijere, A., Eds.; Wiley-Interscience: New York, 2002; p 493.
- Sonogashira, K. J. Organomet. Chem. 2002, 653, 46.
- Rossi, R.; Carpita, A.; Bellina, F. Org. Prep. Proced. Int. 1995, 27, 127.
- Sonogashira, K. InComprehensiVe Organic Synthesis; Trost, B. M., Fleming, I., Eds.;Pergamon: Oxford, 1991; Vol. 3, p 521.
- Handbook of Organopalladium Chemistry for Organic Synthesis; Negishi, E., de Meijere, A., Eds.; Wiley: New York, 2002.
- Soheili, A.; Albaneze-Walker, J.; Murry, J. A.; Dormer, P. G.; Hughes, D. L. Org. Lett. 2003, 5, 4191.
- Choudary, B. M.; Madhi, S.; Kantam, M. L.; Sreedhar, B.; Iwasawa, Y. J. Am. Chem. Soc. 2004, 126, 2292.
- Posset, T.; Bumel, J. J. Am. Chem. Soc. 2006, 128, 8394.
- Niemela, E. H.; Lee, A. F.; Fairlamb, I. J. S. Tetrahedron Lett. 2004, 45, 3593.
- Stambuli, J. P.; Buhl, M.; Hartwig, J. F. J. Am. Chem. Soc. 2002, 124, 9346.
- Bertus, P.; Fecourt, F.; Bauder, C.; Pale, P. New J. Chem. 2004, 28, 12.
- Letinois-Halbes, U.; Pale, P.; Berger, S. J. Org. Chem. 2005, 70, 9185.
- Karak, M.; Barbosa, L. A.; Hargaden, G. C. RSC Adv., 2014, 4, 53442
- Deng, X.; Mani, N. S. Org. Lett. 2006, 8, 269.
- Name Reactions for Homologation, Part 1; Li, J. J., Eds.; Wiley: New York, 2009.
- Tetrahedron, S.; Lopez, J. C.Valverde, A.; Barrio, A.; PedregosaGomez, A. M.; Lett. 2004, 45, 6307.
- Tykwinski, R. R. Angew. Chem. Int. Ed. 2003, 42, 1566.
- Barrios-Landeros, F.; Hartwig, J. F. J. Am. Chem. Soc. 2005, 127, 6944.
- Gelman, D.; Buchwald, S. L. Angew. Chem., Int. Ed. 2003, 42, 5993.
- Adjabeng, G.; Brenstrum, T.; Frampton, C. S.; Robertson, A. J.; Hillhouse, J.; McNulty, J.; Capretta, A. J. Org. Chem. 2004, 69, 5082.
- Brenstrum, T.; Clattenburg, J.; Britten, J.; Zavorine, S.; Dyck, J.; Robertson, A. J.; McNully, J.; Capretta, A. Org. Lett. 2006, 8, 103.
- Amatore, C.; Jutand, A. J. Organomet. Chem. 1999, 576, 254.
- Buchmeiser, M. R.; Schareina, T.; Kempe, R.; Wurst, K. J. Organomet. Chem. 2001, 634, 39.
- Park, S. B.; Alper, H. Chem. Commun. 2004, 1306.
- Gossage, R. A.; Jenkins, H. A.; Yadav, P. N. Tetrahedron Lett. 2004, 45, 7689.
- Eisnor, C. R.; Gossage, R. A.; Yadav, P. N. Tetrahedron 2006, 62, 3395.
- Gil-Moltó, J.; Nójera, C. Eur. J. Org. Chem. 2005, 19, 4073.
- Eisnor, C. R.; Gossage, R. A.; Yadav, P. N. Tetrahedron 2006, 62, 3395.
- Mas-Marzá, E.; Segarra, A. M.; Claver, C.; Peris, E.; Fernández, E. Tetrahedron Lett. 2003, 44, 6595.
- Herrmann, W. A.; Reisinger, C.-P.; Spiegler, M. J. Organomet. Chem. 1998, 557, 93.
- McGuinness, D. S.; Cavell, K. J. Organometallics 2000, 19, 741.
- Elangovan, A.; Wang, Y.-H.; Ho, T.-I. Org. Lett. 2003, 5, 1841.
- Gil-Moltó, J.; Nájera, C. Adv. Synth. Catal. 2006, 348, 1874
Contributors and Attributions
- Noalle Fellah and Xiaolong Zhu