
12.2: Organometallic Catalysts
- Page ID
- 283109

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Organometallic Compounds as Hydrogenation Catalysts
Migratory insertions play an important role in catalysis.
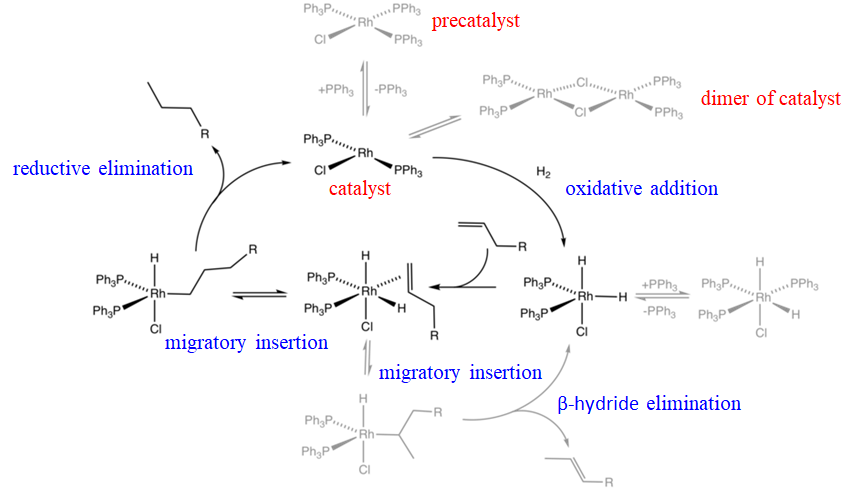
For example a Rh-catalyst called the Wilkinson catalyst is an effective hydrogenation catalyst for olefins. The mechanism of the hydrogenation involves a combination of oxidative additions, olefin migratory insertions, and reductive eliminations (Fig. 12.2.2). Wilkinson’s catalyst is the square planar chloro tris(triphenylphosphine) rhodium(I) complex. This molecule is actually a precatalyst that becomes the actual catalyst when it statistically loses a triphenylphosphine ligand producing chloro bis(triphenylphosphine) rhodium(I). The loss of this ligand is a reversible reaction, and thus the catalyst is in chemical equilibrium with the precatalyst. The actually catalyst is in a second chemical equilibrium with its dimer. The chloro bis(triphenylphosphine) rhodium(I) catalyst can undergo an oxidative addition in the presence of hydrogen to form a trigonal bipyramidal chlorodihydrido bis(triphenylphosphine) (III) rhodium complex. This species is in chemical equilibrium with an octahedral chlorodihydrido tris(triphenyl phosphine) rhodium(III) species that can form due to the presence of free triphenylphosphine ligands in the system. The trigonal bipyramidal species can then add an olefin that binds side-on to the Rh. Because the olefin is in cis-position to the hydride ligand it can undergo an olefin insertion. The Rh-C bond can either form with the first or the second carbon in the carbon chain of the olefin, giving a linear and a branched alkyl complex, respectively. The branched complex can undergo a β-hydride elimination thereby reforming the trigonal bipyramidal Rh-complex, and an olefin. This reaction is a side-reaction because the branched alkyl complex is sterically more crowded than the linear complex. The linear alkyl Rh complex can undergo a reductive elimination to form the linear alkane and the RhCl(PPh3)2 catalyst. This completes the catalytic cycle, and a new cycle can start.
Ziegler-Natta Polymerization
Another example of an organometallic catalytic reaction is the Ziegler-Natta olefin polymerization. This reaction is of high industrial importance for the production of olefins like polyethylene. There are both heterogeneous and homogeneous Ziegler-Natta catalysts. The mechanism for the homogeneous catalysts is generally well understood. Homogeneous catalysts are typically metallocene catalysts.
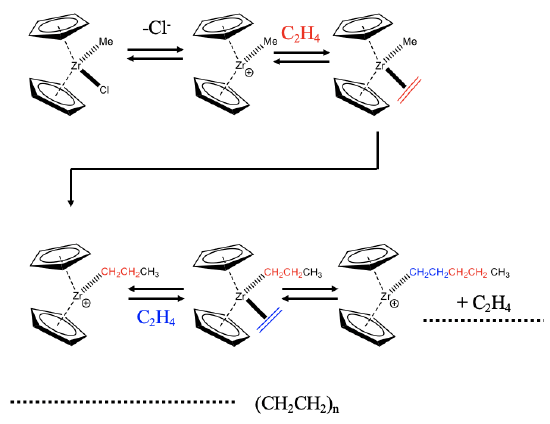
An example of a zirconium-based catalyst is shown (Fig. 12.2.3). The catalyst is a coordinatively unsaturated complex cation with two cyclopentadienyl rings and a methyl group. The catalyst is formed from its precatalyst, a neutral molecule with an additional chloro ligand. The catalyst oxidatively adds an olefin like an ethylene molecule to the coordinatively unsaturated site. This step is followed an olefin insertion step that produces a propyl group. The migratory insertion leads to the formation of a vacant site, that can be re-oocupied by another ethylene molecule. This molecule can insert into the propyl chain thereby prolonging the propyl chain to a pentyl chain. The olefin insertion step generates another vacant site that can be reoccupied by a new ethylene molecule. Repeating the catalytic cycle many times eventually leads to polyethylene.
Catalytic Olefin Hydroformylation
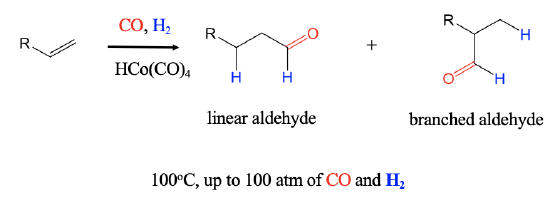
A further important industrial reaction is the catalytic hydroformylation reaction, also known as oxo-process. It was discovered in 1938 by Otto Roelen at BASF. In the hydroformylation reaction an H atom and a formyl group are added to an alkene to form aldehydes. The reaction can produce both branched an linear aldehydes from terminal alkenes, CO, and H2 using a carbonyl hydrides such as HCo(CO)4 as a catalyst. The reaction is performed at about 100°C at a pressure of up to 100 atm.
Mechanism of the Catalytic Olefin Hydroformylation by HCo(CO)4
How does the hydroformylation work mechanistically?
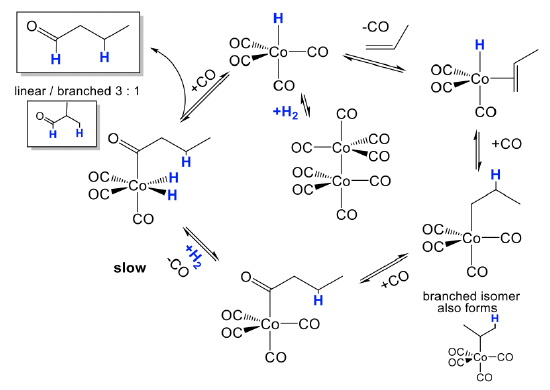
The mechanism is illustrated for the hydroformylation of propene (Fig. 12.2.5). The actual catalyst HCo(CO)4 is first formed from its precatalyst Co2(CO)8 in the presence of H2 in a dinuclear oxidative addition reaction. The catalyst can undergo a substitution reaction in which a CO ligand is replaced by the olefin that binds side-on to the cobalt. This species can then undergo a migratory olefin insertion reaction. This leads to a mixture of linear and branched alkyl groups attached to the Co. A new CO ligand can add to the vacant site. The alkyl group can then insert into a carbonyl group in another migratory insert step, and the vacant site can be reoccupied by a new CO molecule. Then, H2 is added in an oxidative addition. This is the slowest and rate-limiting step in the catalytic cycle. From the addition product the aldehyde can then be eliminated in a reductive elimination reaction. Addition of CO regenerates the catalyst, and the catalytic cycle can begin again.
Hydrocarbonylations
After the hydroformylation, a number of other hydrocarbonylations were developed, and industrially deployed.
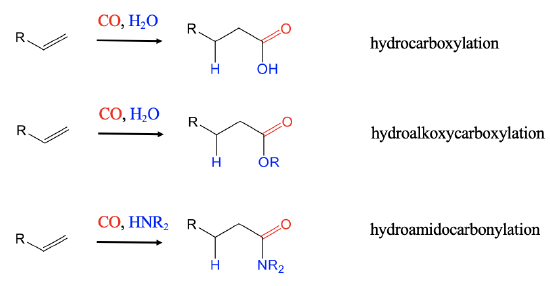
When hydrogen is replaced by H2O hydrocarboxylations of alkenes lead to carboxylic acids (Fig. 12.2.6). With an alcohol instead of H2 hydroalkoxycarbonylcations lead to esters. The employment of amines instead of H2 leads to amides in hydroamidocarbonylation reactions.
The Monsanto Acetic Acid Process
Another carbonylation reaction involving an organometallic catalyst is the Monsanto acetic acid process. It has been introduced by Monsanto in the 1970s for the industrial production of acetic acid from methanol. The reaction involves dual catalysis with HI and [RhI2(CO)2]- as a co-catalysts. How does this reaction work?
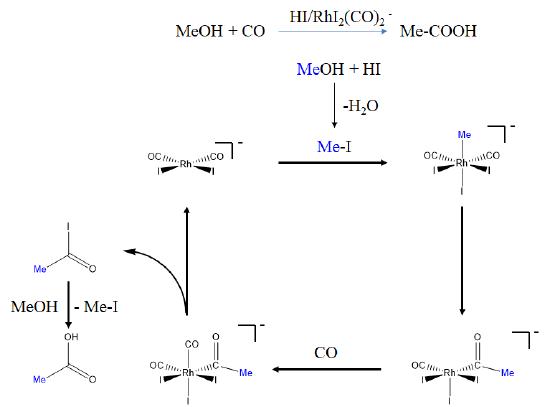
In the first step methanol reacts with HI to form methyl iodide. The methyl iodide then reacts with the Rh-catalyst in an oxidative addition reaction in which a methyl and an iodo group are added in trans-fashion to the square-planar Rh-complex to give an octahedral complex. The octahedral complex then undergoes a migratory insertion reaction with CO producing an acyl group and a vacant site. A CO molecule can then add to the vacant site. The acetyl iodide can then be eliminated in a reductive elimination to reform the Rh-catalyst thereby closing the catalytic cycle. The acetyl iodide can then react with methanol to form new methyl iodide and acetic acid. The methyl iodide can start a new catalytic cycle with the Rh-catalyst.
Olefin Metathesis
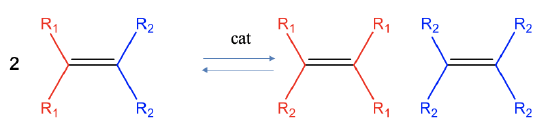
Olefin metathesis is a reaction which allows to cut and rearrange C=C double bonds in olefins to make new olefins (Fig. 12.2.8). Formally, the carbon-carbon bond of the reactant is cleaved homoleptically and the two carbene fragments are combined in a different way. This reaction is typically an equilibrium reaction, and neither the reactants nor the products are clearly favored. This reaction is catalyzed by molybdenum arylamido carbene complexes or ruthenium carbene complexes.
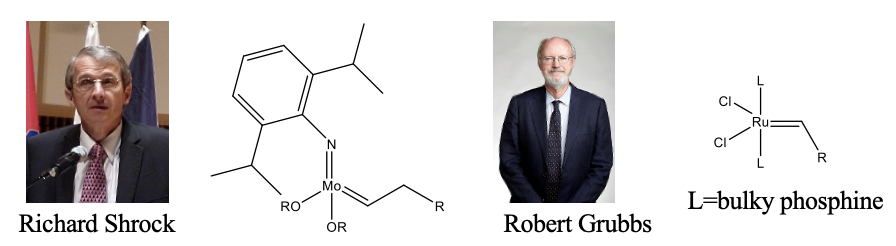
The former are called Shrock catalysts, and the latter Grubbs catalysts named after their discoverers Richard Shrock and Robert Grubbs who received the Nobel prize for Chemistry in 2005 (Fig. 12.2.9). The Schrock catalysts are more active, but also very sensitive to air and water. The Grubbs catalysts, while less active, are less sensitive to air and water.
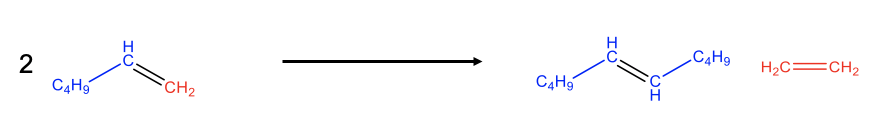
Olefin metathesis often allows for simpler preparation of olefins compared to other methods. Olefin metathesis is particularly powerful when one olefin product is gaseous because then it can be quite easily removed from the chemical equilibrium by purging. This drives the chemical equilibrium to the right side. An example is the preparation of 5-decene from 1-hexene. Cleavage of the C=C double bond in the hexene leads to C5 and C1 carbene fragments (Fig. 12.2.10). The two C1 fragments can combine to form ethylene and the two C5 fragments combine to 5-decene. The ethylene is volatile and can be purged from the reaction system thereby driving the chemical reaction to the right side.
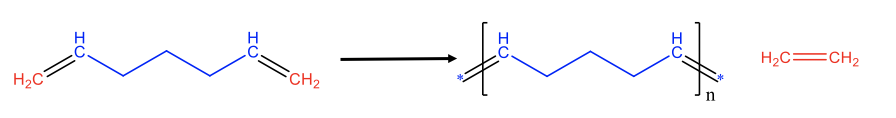
The same principles can also be applied to produce polymers from dienes with two terminal C=C double bonds at the chain ends. This is called acylic diene metathesis (ADMET), Fig. 12.2.11. For instance the cleavage of the two terminal double bonds in a diene with seven C atoms leads to C1 and C5 fragments. The C1 fragments can combine to form ethylene, and the C5 fragments can combine to make an unsaturated polymer of the type [CH(CH2)3CH]n. Again, the reaction can be driven to the right side by removing the gaseous ethylene from the reaction mixture through purging.
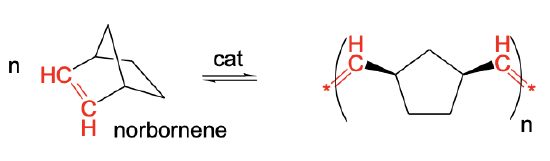
Another variation of olefin metathesis is ring-opening metathesis polymerization (ROMP). It allows to make polymers from strained cycloolefins, for example norbornene. The reaction driving force is the relief of the strain. Because the strain is removed in the polymer, the chemical equilibrium lies far on the right side. The reaction product in norbornene is a polymer with 5-membered rings that are interconnected by ethylene -CH=CH- units (Fig. 12.2.12).
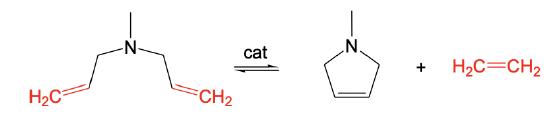
The opposite of ROMP is ring-closure metathesis (RCM). RCM allows for the preparation of unstrained rings with C=C double bonds from dienes with C=C double bonds that are five or six carbon atoms apart. This distance is suitable to produce unstrained rings. In the shown example a five-membered ring with a C=C double bond is formed from a diene with terminal C=C double bonds that are five atoms apart.
The Mechanism of Olefin Metathesis
What is the mechanism of olefin metathesis?
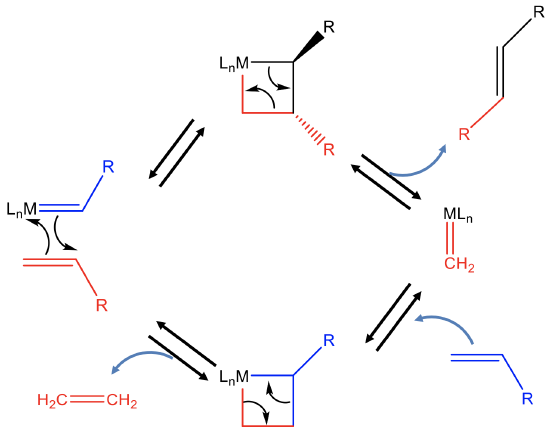
In the first step, the alkene adds to the the carbene fragment of the catalyst in a 2+2 cycloaddition reaction to produce an unstable intermediate with a highly strained four-membered ring (Fig. 12.2.14). This four membered ring can open to produce the first new alkene product R-CH=CH-R and a metal carbene species. This metal carbene can react with another reactant olefin to form another highly stained 4-ring intermediate via a 2+2 cycloaddition reaction. This ring can then reopen again to produce the second alkene metathesis product, in this case ethylene, and the original catalyst. The regenerated catalyst can then start a new catalytic cycle.
Dr. Kai Landskron (Lehigh University). If you like this textbook, please consider to make a donation to support the author's research at Lehigh University: Click Here to Donate.