
6.5: Regulatory Frameworks
- Page ID
- 294576

6.5. Regulatory Frameworks
Regulatory frameworks
Authors: Charles Bodar and Joop de Knecht
Reviewers: Kees van Gestel
Learning objectives:
You should be able to
- explain how the potential environmental risks of chemicals are legally being controlled in the EU and beyond
- mention the different regulatory bodies involved in the regulation of different categories of chemicals
- explain the purpose of the Classification, Labelling and Packaging (CLP) approach and its difference with the risk assessment of chemicals
Keywords: chemicals, environmental regulations, hazard, risk
Introduction
There is no single, overarching global regulatory framework to manage the risks of all chemicals. Instead, different regulations or directives have been developed for different categories of chemicals. These categories are typically related to the usage of the chemicals. Important categories are industrial chemicals (solvents, plasticizers, etc.), plant protection products, biocides and human and veterinary drugs. Some chemicals may belong to more than one category. Zinc, for example, is used in the building industry, but it also has biocidal applications (antifouling agent) and zinc oxide is used as a veterinary drug. In the European Union, each chemical category is subject to specific regulations or directives providing the legal conditions and requirements to guarantee a safe production and use of chemicals. A key element of all legal frameworks is the requirement that sufficient data on a chemical should be made available. Valid data on production and identity (e.g. chemical structure), use volumes, emissions, environmental fate properties and the (eco)toxicity of a chemical are the essential building blocks for a sound assessment and management of environmental risks. Rules for the minimum data set that should be provided by the actors involved (e.g. producers or importers) are laid down in various regulatory frameworks. With this data, both hazard and risk assessments can be carried out according to specified technical guidelines. The outcome of the assessment is then used for risk management, which is focused on minimizing any risk by taking measures, ranging from requests for additional data to restrictions on the particular use or a full-scale ban of a chemical.
REACH
REACH is a regulation of the European Union, adopted to improve the protection of human health and the environment from the risks that can be posed by chemicals, while enhancing the competitiveness of the EU chemicals industry. REACH stands for Registration, Evaluation, Authorisation and Restriction of Chemicals. The REACH regulation entered into force on 1st June 2007 to streamline and improve the former legislative frameworks on new and existing chemical substances. It replaced approximately forty community regulations and directives by one single regulation.
REACH establishes procedures for collecting and assessing information on the properties, hazards and risks of substances. REACH applies to a very broad spectrum of chemicals: from industrial to household applications, and very much more. It requires that EU manufacturers and importers register their chemical substances if produced or imported in annual amounts of > 1 tonne, unless the substance is exempted from registration under REACH. At quantities of > 10 tonnes, the manufacturers, importers, and down-stream users are responsible to show that their substances do not adversely affect human health or the environment.
The amount of standard information required to show safe use depends on the quantity of the substance that is manufactured or imported. Before testing on vertebrate animals like fish and mammals, the use of alternative methods must be considered. The European Chemical Agency (ECHA) coordinates and facilitates the REACH program. For production volumes above 10 tonnes per year, industry has to prepare a risk assessment, taking into account all risk management measures envisaged, and document this in a chemical safety assessment (CSA). A CSA should include an exposure assessment, hazard or dose-response assessment, and a risk characterization showing risks ratios below 1.0, i.e. safe use (see sections on REACH Human and REACH Eco).
Classification, Labelling and Packaging (CLP)
The EU CLP regulation requires manufacturers, importers or downstream users of substances or mixtures to classify, label and package their hazardous chemicals appropriately before placing them on the market. When relevant information (e.g. ecotoxicity data) on a substance or mixture meets the classification criteria in the CLP regulation, the hazards of a substance or mixture are identified by assigning a certain hazard class and category. An important CLP hazard class is 'Hazardous to the aquatic environment', which is divided into categories based on the criteria, for example, Category Acute 1, representing the most (acute) toxic chemicals (LC50/EC50 ≤ 1 mg/L). CLP also sets detailed criteria for the labelling elements, such as the well-known pictograms (Figure 1).

Plant protection products regulation
Plant protection products (PPPs) are pesticides that are mainly used to keep crops healthy and prevent them from being damaged by disease and infestation. They include among others herbicides, fungicides, insecticides, acaricides, plant growth regulators and repellents (see section on Crop Protection Products). PPPs fall under the EU Regulation (EC) No 1107/2009 which determines that PPPs cannot be placed on the market or used without prior authorization. The European Food and Safety Authority (EFSA) coordinates the EU regulation on PPPs.
Biocides regulation
The distinction between biocides and PPP is not always straightforward, but as a general rule of thumb the PPP regulation applies to substances used by farmers for crop protection while the biocides regulation covers all other pesticide applications. Different applications of the same active ingredient, one as a PPP and the other as a biocide, may thus fall under different regulations. Biocides are used to protect humans, animals, materials or articles against harmful organisms like pests or bacteria, by the action of the active substances contained in the biocidal product. Examples of biocides are antifouling agents, preservatives and disinfectants.
According to the EU Biocidal Products Regulation (BPR), all biocidal products require an authorization before they can be placed on the market, and the active substances contained in the biocidal product must be previously approved. The European Chemical Agency (ECHA) coordinates and facilitates the BPR. More or less similar to other legislations, for biocides the environmental risk assessment is mainly performed by comparing compartmental concentrations (PEC) with the concentration below which unacceptable effects on organisms will most likely not occur (PNEC).
Veterinary and human pharmaceuticals regulation
Since 2006, EU law requires an environmental risk assessment (ERA) for all new applications for a marketing authorization of human and veterinary pharmaceuticals. For both products, guidance documents have been developed for conducting an ERA based on two phases. The first phase estimates the exposure of the environment to the drug substance. Based on an action limit the assessment may be terminated. In the second phase, information about the fate and effects in the environment is obtained and assessed. For conducting an ERA a base set, including ecotoxicity data, is required. For veterinary medicines, the ERA is part of a risk-benefit analysis, in which the positive therapeutic effects are weighed against any environment risks, whereas for human medicines the environmental concerns are excluded from the risk-benefit analysis. The European Medicines Agency (EMA) is responsible for the scientific evaluation, supervision and safety monitoring of medicines in the EU.
Harmonization of testing
Testing chemicals is an important aspect of risk assessment, e.g. testing for toxicity, for degradation or for a physicochemical property like the Kow (see Chapter 3). The outcome of a test may vary depending on the conditions, e.g. temperature, test medium or light conditions. For this reason there is an incentive to standardize the test conditions and to harmonize the testing procedures between agencies and countries. This would also avoid duplication of testing, and leading to a more efficient and effective testing system.
The Organization for Economic Co-operation and Development (OECD) assists its member governments in developing and implementing high-quality chemical management policies and instruments. One of the key activities to achieve this goal is the development of harmonized guidelines to test and assess the risks of chemicals leading to a system of mutual acceptance of chemical safety data among OECD countries. The OECD also developed Principles of Good Laboratory Practice (GLP) to ensure that studies are of sufficient quality and rigor and are verifiable. The OECD also facilitates the development of new tools to obtain more safety information and maintain quality while reducing costs, time and animal testing, such as the OECD QSAR toolbox.
What are the major categories of chemicals for which regulatory frameworks are present controlling the environmental risks of these chemicals?
Is a chemical producer in the EU allowed to put a new chemical on the market without a registration or authorization?
Is the CLP regulation based on the hazard, exposure and/or risk of chemicals?
The amount of minimum information that is required under REACH for making a proper hazard and risk assessment is dependent on what? And what do you think is the rationale behind it?
Name three European agencies or authorities that are coordinating important regulatory frameworks in the EU?
6.5.1. REACH human
Authors: Theo Vermeire
Reviewers: Tim Bowmer
Learning objective:
You should be able to:
- outline how human risk assessment of chemicals is performed under REACH;
- explain the regulatory function of human risk assessment in REACH.
Keywords: REACH, chemical safety assessment, human, RCR, DNEL, DMEL
Human risk assessment under REACH
The REACH Regulation aims to ensure a high level of protection of human health and the environment, including the promotion of alternative methods for assessment of hazards of substances, as well as the free circulation of substances on the internal market while enhancing competitiveness and innovation. Risk assessment under REACH aims to realize such a level of protection for humans that the likelihood of adverse effects occurring is low, taking into account the nature of the potentially exposed population (including sensitive groups) and the severity of the effect(s). Industry therefore has to prepare a risk assessment (in REACH terminology: chemical safety assessment, CSA) for all relevant stages in the life cycle of the chemical, taking into account all risk management measures envisaged, and document this in the chemical safety report (CSR). Risk characterization in the context of a CSA is the estimation of the likelihood that adverse effect levels occur due to actual or predicted exposure to a chemical. The human populations considered, or protection goals, are workers, consumers and humans exposed via the environment. In risk characterization, exposure levels are compared to reference levels to yield "risk characterization ratios" (RCRs) for each protection goal. RCRs are derived for all endpoints (e.g. skin and eye irritation, sensitization, repeated dose toxicity) and time scales. It should be noted that these RCRs have to be derived for all stages in the life-cycle of a compound.
Environmental exposure assessment for humans
Humans can be exposed through the environment directly via inhalation of indoor and ambient air, soil ingestion and dermal contact, and indirectly via food products and drinking water (Figure 1). REACH does not consider direct exposure via soil.

In the REACH exposure scenario, assessment of human exposure through the environment can be divided into three steps:
- Determination of the concentrations in intake media (air, soil, food, drinking water);
- Determination of the total daily intake of these media;
- Combining concentrations in the media with total daily intake (and, if necessary, using a factor for bioavailability through the route of uptake concerned).
A fourth step may be the consideration of aggregated exposure taking into account exposure to the same substance in consumer products and at the workplace. Moreover, there may be similar substances, acting via the same mechanism of action, that may have to be considered in the exposure assessment, for instance, as a worst case, by applying the concept of dose or concentration addition.
The section on Environmental realistic scenarios (PECs) - Human explains the concept of exposure scenarios and how concentrations in environmental compartments are derived.
Hazard identification and dose-response assessment
The aim of hazard identification is to classify chemicals and to select key data for the dose-response assessment to derive a safe reference level, which in REACH terminology is called the DNEL (Derived No Effect Level) or DMEL (Derived Minimal Effect Level). For human end-points, a distinction is made between substances considered to have a threshold for toxicity and those without a threshold. For threshold substances, a No-Observed-Adverse Effect Level (NOAEL) or Lowest-Observed-Adverse-Effect Level (LOAEL) is derived, typically from toxicity studies with laboratory animals such as rats and mice. Alternatively a Benchmark Dose (BMD) can be derived by fitting a dose-response model to all observations. These toxicity values are then extrapolated to a DNEL using assessment factors to correct for uncertainty and variability. The most frequently used assessment factors are those for interspecies differences and those for intraspecies variability (see section on Setting safe standards). Additionally, factors can be applied to account for remaining uncertainties such as those due to a poor database.
For substances considered to exert their effect by a non-threshold mode of action, especially mutagenicity and carcinogenicity, it is generally assumed, as a default assumption, that even at very low levels of exposure residual risks cannot be excluded. That said, recent progress has been made on establishing scientific, 'health-based' thresholds for some genotoxic carcinogens. For non-threshold genotoxic carcinogens it is recommended to derive a DMEL, if the available data allow. A DMEL is a cancer risk value considered to be of very low concern, e.g. a 1 in a million tumour risk after lifetime exposure to the chemical and using a conservative linear dose-response model. There is as yet no EU-wide consensus on acceptable levels of cancer risk.
Risk characterization
Safe use of substances is demonstrated when:
• RCRs are below one, both at local and regional level. For threshold substances, the RCR is the ratio of the estimated exposure (concentration or dose) and the DNEL; for non-threshold substances the DMEL is used.
• The likelihood and severity of an event such as an explosion occurring due to the physicochemical properties of the substance as determined in the hazard assessment is negligible.
A risk characterization needs to be carried out for each exposure scenario (see Section on Environmental realistic scenarios (PECs) - Human) and human population. The assessment consists of a comparison of the exposure of each human population known to be or likely to be exposed with the appropriate DNELs or DMELs and an assessment of the likelihood and severity of an event occurring due to the physicochemical properties of the substance.
|
Example of a deterministic assessment (Vermeire et al., 2001) Exposure assessmentBased on an emission estimation for processing of dibutylphthalate (DBP) as a softener in plastics, the concentrations in environmental compartments were estimated. Based on modelling as schematically presented in Figure 1, the total human dose was determined to be 93 ug.kg bw-1.
Effects assessmentThe total dose should be compared to a DNEL for humans. DBP is not considered a genotoxic carcinogen but is toxic to reproduction and therefore the risk assessment is based on endpoints assumed to have a threshold for toxicity. The lowest NOAEL of DBP was observed in a two-generation reproduction test in rats and at the lowest dose-level in the diet (52 mg.kgbw-1.d-1 for males and 80 mg.kgbw-1.d-1 for females) a reduced number of live pups per litter and decreased pup weights were seen in the absence of maternal toxicity. The lowest dose level of 52 mg.kgbw-1.d-1 was chosen as the NOAEL. The DNEL was derived by the application of an overall assessment factor of 1000, accounting for interspecies differences, human variability and uncertainties due to a non-chronic exposure period. Risk characterisationThe deterministic estimate of the RCR would be based on the deterministic exposure estimate of 0.093 mg.kgbw-1.d-1 and the deterministic DNEL of 0.052 mg.kgbw-1.d-1. The deterministic RCR would then be 1.8, based on the NOAEL. Since this is higher than one, this assessment indicates a concern, requiring a refinement of the assessment or risk management measures. |
Additional reading
Van Leeuwen C.J., Vermeire T.G. (Eds.) (2007) Risk assessment of chemicals: an introduction. Springer, Dordrecht, The Netherlands, ISBN 978-1-4020-6102-8 (e-book), https://doi.org/10.1007/978-1-4020-6102-8.
Vermeire, T., Jager, T., Janssen, G., Bos, P., Pieters, M. (2001) A probabilistic human health risk assessment for environmental exposure to dibutylphthalate. Journal of Human and Ecological Risk Assessment 7, 1663-1679.
Uncertainty happens! It is inherent to risk assessment. Where, in your view, are the greatest sources of uncertainty in the process of risk assessment?
Are there risks identified in the example for humans indirectly exposed via the environment? To what extent are these potential or realistic risks by asking yourself the following questions:
• Do you have relevant toxicological and exposure data ?
• Are these fixed values or not?
• How relevant or adverse are the toxicological effects observed?
• Were appropriate assessment factors used?
What would you recommend as a strategy to reduce the identified risks sufficiently?
6.5.2. REACH environment
Author: Joop de Knecht
Reviewers: Watze de Wolf
Keywords: REACH, European chemicals regulation
Introduction
REACH establishes procedures for collecting and assessing information on the properties, hazards and risks of substances. At quantities of > 10 tonnes, the manufacturers, importers, and down-stream users must show that their substances do not adversely affect human health or the environment for the uses and operational conditions registered. The amount of standard information required to show safe use depends on the quantity of the substance that is manufactured or imported. This section explains how risks to the environment are assessed in REACH.
Data requirements
As a minimum requirement, all substances manufactured or imported in quantities of 1 tonne or more need to be tested in acute toxicity tests on Daphnia and algae, while also information should be provided on biodegradability (Table 1). Physical-chemical properties relevant for environmental fate assessment that have to be provided at this tonnage level are water solubility, vapour pressure and octanol-water partition coefficient. At 10 tonnes or more, this should be supplemented with an acute toxicity test on fish and an activated sludge respiration inhibition test. At this tonnage level, also an adsorption/desorption screening and a hydrolysis test should be performed. If the chemical safety assessment, performed at 100 tonnes or more in case a substance is classified based on hazard information, indicates the need to investigate further the effects on aquatic organisms, the chronic toxicity on these aquatic species should be determined. If the substance has a high potential for bioaccumulation (for instance a log Kow > 3), also the bioaccumulation in aquatic species should be determined. The registrant should also determine the acute toxicity to terrestrial species or, in the absence of these data, consider the equilibrium partitioning method (EPM) to assess the hazard to soil organisms. To further investigate the fate of the substance in surface water, sediment and soil, simulation tests on its degradation should be conducted and when needed further information on the adsorption/desorption should be provided. At 1000 tonnes or more, chronic tests on terrestrial and sediment-living species should be conducted if further refinement of the safety assessment is needed. Before testing vertebrate animals like fish and mammals, the use of alternative methods and all other options must be considered to comply with the regulations regarding (the reduction of) animal testing.
Table 1 Required ecotoxicological and environmental fate information as defined in REACH
|
1-10 t/y |
|
|
10-100 t/y |
|
|
100-1000 t/y |
|
|
≥ 1000 t/y |
|
Safety assessment
For substances that are classified based on hazard information the registrant should assess the environmental safety of a substance, by comparing the predicted environmental concentration (PEC) with the Predicted No Effect Concentration (PNEC), resulting in a Risk Characterisation Ratio (RCR=PEC/PNEC). The use of the substance is considered to be safe when the RCR <1.
Chapter 16 of the ECHA guidance offers methods to estimate the PEC based on the tonnage, use and operational conditions, standardised through a set of use descriptors, particularly the Environmental Release Categories (ERCs). These ERCs are linked to conservative default release factors to be used as a starting point for a first tier environmental exposure assessment. When substances are emitted via waste water, the physical-chemical and fate properties of the chemical substance are then used to predict its behaviour in the Wastewater Treatment Plant (WWTP). Subsequently, the release of treated wastewater is used to estimate the concentration in fresh and marine surface water. The concentration in sediment is estimated from the PEC in water and experimental or estimated sediment-water partitioning coefficient (Kpsed). Soil concentrations are estimated from deposition from air and the application of sludge from an WWTP. The guidance offers default values for all relevant parameters, thus a generic local PEC can be calculated and considered applicable to all local emissions in Europe, although the default values can be adapted to specific conditions if justified. The local risk for wide-dispersive uses (e.g. from consumers or small, non- industrial companies) is estimated for a default WWTP serving 10,000 inhabitants. In addition, a regional assessment is conducted for a standard area, a region represented by a typical densely populated EU area located in Western Europe (i.e. about 20 million inhabitants, distributed in a 200 x 200 km2 area). For calculating the regional PECs, a multi-media fate-modelling approach is used (e.g. the SimpleBox model; see Section on Multicompartment fate modelling). All releases to each environmental compartment for each use, assumed to constitute a constant and continuous flux, are summed and averaged over the year and steady-state concentrations in the environmental compartments are calculated. The regional concentrations are used as background concentrations in the calculation of the local concentrations.
The PNEC is calculated using the lowest toxicity value and an assessment factor (AF) related to the amount of information (see section Setting safe standards or chapter 10 of the REACH guidance. If only the minimum set of aquatic acute toxicity data is available, i.e. LC50s or EC50s for algae, daphnia and fish, a default value of 1000 is used. When one, two or three or more long-term tests are available, a default AF of 100, 50 and 10 is applied to No Observed Effect Concentrations (NOECs), respectively. The idea behind lowering the AF when more data become available is that the amount of uncertainty around the PNEC is being reduced.
In the absence of ecotoxicological data for soil and/or sediment-dwelling organisms, the PNECsoil and/or PNECsed may be provisionally calculated using the EPM. This method uses the PNECwater for aquatic organisms and the suspended matter/water partitioning coefficient as inputs. For substances with a log Kow >5 (or with a corresponding log Kp value), the PEC/PNEC ratio resulting from the EPM is increased by a factor of 10 to take into account possible uptake through the ingestion of sediment. If the PEC/PNEC is greater than 1 a sediment test must be conducted. If one, two or three long-term No Observed Effect Concentrations (NOECs) from sediment invertebrate species representing different living and feeding conditions are available, the PNEC can be derived using default AFs of 100, 50 and 10, respectively.
For data rich chemicals, the PNEC can be derived using Species Sensitivity Distributions (SSD) or other higher-tier approaches.
Who is responsible within the EU that industrial chemicals do not pose a risk for the environment?
In which circumstances will an environmental risk be identified?
Describe how the ecotoxicogical safety levels (PNECs) for the aquatic environment are derived depending on the ecotoxicological information available?
6.5.3. Pesticides (EFSA)
under review
6.5.4. Environmental Risk Assessment of Pharmaceuticals in Europe
Author: Gerd Maack
Reviewers: Ad Ragas, Julia Fabrega, Rhys Whomsley
Learning objectives:
You should be able to
- Explain the philosophy and objective of the environmental risk assessment of pharmaceuticals.
- mention the key aspects of the tiered approach of the assessment
- identify the exposure routes for human and veterinary medicinal products and should know the respective consequences in the assessment
Keywords: Human pharmaceuticals, veterinary pharmaceuticals, environmental impact, tiered approach
Introduction
Pharmaceuticals are a crucial element of modern medicine and confer significant benefits to society. About 4,000 active pharmaceutical ingredients are being administered worldwide in prescription medicines, over-the-counter medicines, and veterinary medicines. They are designed to be efficacious and stable, as they need to pass different barriers i.e. skin, the gastrointestinal system (GIT), or even the blood-brain barrier before reaching the target cells. Each target system has a different pH and different lipophilicity and the GIT is in addition colonised with specific bacteria, specialized to digest, dissolve and disintegrate organic molecules. As a consequence of this stability, most of the pharmaceutical ingredients are stable in the environment as well and could cause effects on non-target organisms.
The active ingredients comprise a variety of synthetic chemicals produced by pharmaceutical companies in both the industrialized and the developing world at a rate of 100,000 tons per year.
While pharmaceuticals are stringently regulated in terms of efficacy and safety for patients, as well as for target animal safety, user and consumer safety, the potential effects on non-target organisms and environmental effects are regulated comparably weakly.
The authorisation procedure requires an environmental risk assessment (ERA) to be submitted by the applicants for each new human and veterinary medicinal product. The assessment encompasses the fate and behaviour of the active ingredient in the environment and its ecotoxicity based on a catalogue of standardised test guidelines.
In the case of veterinary pharmaceuticals, constraints to reduce risk and thus ensure safe usage can be stipulated in most cases. In the case of human pharmaceuticals, it is far more difficult to ensure risk reduction through restriction of the drug's use due to practical and ethical reasons. Because of their unique benefits, a restriction is not reasonable. This is reflected in the legal framework, as a potential effect on the environment is not included in the final benefit risk assessment for a marketing authorisation.
Exposure pathways
Human pharmaceuticals
Human pharmaceuticals enter the environment mainly via surface waters through sewage systems and sewage treatment plants. The main exposure pathways are excretion and non-appropriate disposal. Typically only a fraction of the medicinal product taken is metabolised by the patients, meaning that the main share of the active ingredient is excreted unchanged into the wastewater system. Furthermore, sometimes the metabolites themselves are pharmacologically active. Yet, no wastewater treatment plant is able to degrade all active ingredients. So medicinal products are commonly found in surface water, to some extent in ground water, and sometimes even in drinking water. However, the concentrations in drinking water are orders of magnitude lower than therapeutic concentrations. An additional exposure pathway for human pharmaceuticals is the spreading of sewage sludge on soil, if the sludge is used as fertilizer on farmland. See for more details the Link "The Drugs We Wash Away: Pharmaceuticals, Drinking Water and the Environment".
Veterinary pharmaceuticals
Veterinary pharmaceuticals on the other hand enter the environment mainly via soil, either indirectly, if the slurry and manure from mass livestock production is spread onto agricultural land as fertiliser, or directly from pasture animals. Moreover, pasture animals might additionally excrete directly into surface water. Pharmaceuticals can also enter the environment via the detour of manure used in biogas plants.

Assessment schemes
Despite the differences mentioned above, the general scheme of the environmental risk assessment of human and veterinary pharmaceuticals is similar. Both assessments start with an exposure assessment. Only if specific trigger values are reached an in-depth assessment of fate, behaviour and effects of the active ingredient is necessary.
Environmental risk assessment of human pharmaceuticals
In Europe, an ERA for human pharmaceuticals has to be conducted according to the Guideline on Environmental Risk Assessment of Medicinal Products for Human Use (EMA 2006). This ERA consists of two phases. Phase I is a pre-screening for estimating the exposure in surface water, and if this Predicted Environmental Exposure Concentration (PEC) does not reach the action limit of 0.01 µg/L, in most cases, the ERA can stop. In case this action limit is reached or exceeded, a base set of aquatic toxicology, and fate and behaviour data need to be supplied in phase II Tier A. A risk assessment, comparing the PEC with the Predicted No Effect Concentration (PNEC), needs to be conducted. If in this step a risk is still identified for a specific compartment, a substance and compartment-specific refinement and risk assessment in Phase II Tier B needs to be conducted (Figure 2).
Phase I: Estimation of Exposure
In Phase I, the PEC calculation is restricted to the aquatic compartment. The estimation should be based on the drug substance only, irrespective of its route of administration, pharmaceutical form, metabolism and excretion. The initial calculation of the PEC in surface water assumes:
- The predicted amount used per capita per year is evenly distributed over the year and throughout the geographic area (Doseai);
- A fraction of the overall market penetration (Fpen), in other words 'how many people will take the medicinal product? Normally a default value of 1% is used;
- The sewage system is the drug's main route of entry into surface water.
The following formula is used to estimate the PEC in surface water:
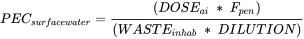
PECsurfacewater = mg/l
DOSEai = Maximum daily dose consumed per capita [mg.inh-1.d-1]
Fpen = Fraction of market penetration (= 1% by default)
WASTEinhab = Amount of wastewater per inhabitant per day (= 200 l by default)
DILUTION = Dilution Factor (= 10 by default)
Three factors of this formula, i.e. Fpen, Wasteinhab and the Dilution Factor, are default values, meaning that the PECsurfacewater in Phase I entirely depends on the dose of the active ingredient. The Fpen can be refined by providing reasonably justified market penetration data, e.g. based on published epidemiological data.
If the PECsurfacewater value is equal to or above 0.01 μg/l (mean dose ≥ 2 mg cap-1 d-1), a Phase II environmental fate and effect analysis should be performed. Otherwise, the ERA can stop. However, in some cases, the action limit may not be applicable. For instance, medicinal substances with a log Kow > 4.5 are potential PBT candidates and should be screened for persistence (P), bioaccumulation potential (B), and toxicity (T) independently of the PEC value. Furthermore, some substances may affect vertebrates or lower animals at concentrations lower than 0.01 μg/L. These substances should always enter Phase II and a tailored risk assessment strategy should be followed which addresses the specific mechanism of action of the substance. This is often true for e.g. hormone active substances (see section on Endocrine disruption). The required tests in a Phase II assessment (see below) need to cover the most sensitive life stage, and the most sensitive endpoint needs to be assessed. This means for instance that for substances affecting reproduction, the organism needs to be exposed to the substance during gonad development and the reproductive output needs to be assessed.
Phase II: Environmental Fate and Effects Analysis
A Phase II assessment is conducted by evaluating the PEC/PNEC ratio based on a base set of data and the predicted environmental concentration from Tier A. If a potential environmental impact is indicated, further testing might be needed to refine PEC and PNEC values in Tier B.
Under certain circumstances, effects on sediment-dwelling organisms and terrestrial environmental fate and effects analysis are also required. Experimental studies should follow standard test protocols, e.g. OECD guidelines. It is not acceptable to use QSAR estimation, modelling and extrapolation from e.g. a substance with a similar mode of action and molecular structure (read across). This is in clear contrast to other regulations like e.g. REACH.
Human pharmaceuticals are used all year round without any major fluctuations and peaks. The only exemption are substances used against cold and influenza. These substances have a clear peak in the consumption in autumn and winter times. In developed countries in Europe and North America, antibiotics display a similar peak as they are prescribed to support the substances used against viral infections. The guideline reflects this exposure scenario and asks explicitly for long-term effect tests for all three trophic levels: algae, aquatic invertebrates and vertebrates (i.e., fish).
In order to assess the physio chemical fate, amongst other tests the sorption behaviour and fate in a water/sediment system should be determined.

If, after refinement, the possibility of environmental risks cannot be excluded, precautionary and safety measures may consist of:
- An indication of potential risks presented by the medicinal product for the environment.
- Product labelling, Summary Product Characteristics (SPC), Package Leaflet (PL) for patient use, product storage and disposal.
Labelling should generally aim at minimising the quantity discharged into the environment by appropriate mitigation measures
Environmental risk assessment of veterinary pharmaceuticals
In the EU, the Environmental Risk Assessment (ERA) is conducted for all veterinary medicinal products. The structure of an ERA for Veterinary Medicinal Products (VMPs) is quite similar to the ERA for Human Medicinal Products. It is also tier based and starts with an exposure assessment in Phase I. Here, the potential for environmental exposure is assessed based on the intended use of the product. It is assumed that products with limited environmental exposure will have negligible environmental effects and thus can stop in Phase I. Some VMPs that might otherwise stop in Phase I as a result of their low environmental exposure, may require additional hazard information to address particular concerns associated with their intrinsic properties and use. This approach is comparable to the assessment of Human Pharmaceutical Products, see above.
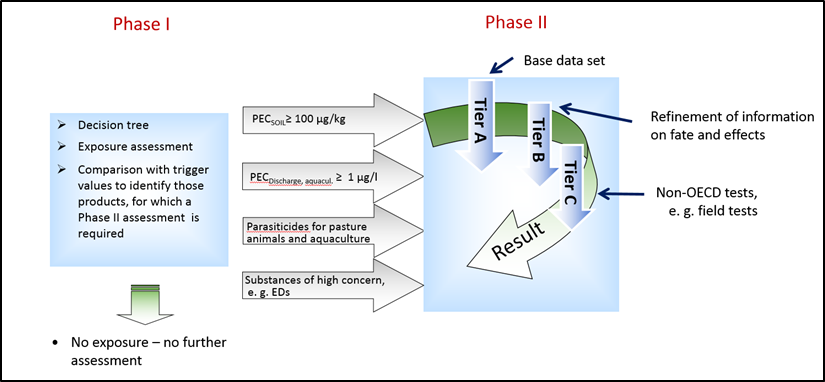
Phase I: Estimation of Environmental Exposure
For the exposure assessment, a decision tree was developed (Figure 3). The decision tree consists of a number of questions, and the answers of the individual questions will conclude in the extent of the environmental exposure of the product. The goal is to determine if environmental exposure is sufficiently significant to consider if data on hazard properties are needed for characterizing a risk. Products with a low environmental exposure are considered not to pose a risk to the environment and hence these products do not need further assessment. However, if the outcome of Phase I assessment is that the use of the product leads to significant environmental exposure, then additional environmental fate and effect data are required. Examples for products with a low environmental exposure are, among others are products for companion animals only and products that result in a Predicted Environmental Concentration in soil (PECsoil) of less than 100 µg/kg, based on a worst-case estimation.

Phase II: Environmental Fate and Effects Analysis
A Phase II assessment is necessary if either the trigger of 100 µg/kg in the terrestrial branch or the trigger of 1 µg/L in the aquatic branch is reached. It is also necessary, if the substance is a parasiticide for food producing animals. A Phase II is also required for substances that would in principle stop in Phase I, but there are indications that an environmental risk at very low concentrations is likely due to their hazardous profile (e.g., endocrine active medicinal products). This is comparable to the assessment for Human Pharmaceutical Products.
For Veterinary Pharmaceutical Products also the Phase II assessment is sub-divided into several Tiers, see Figure 4. For Tier A, a base set of studies assessing the physical-chemical properties, the environmental fate, and effects of the active ingredient is necessary. For Tier A, acute effect tests are suggested, assuming a more peak like exposure scenario due to e.g. applying manure and dung on fields and meadows, in contrast to the permanent exposure of human pharmaceuticals. If for a specific trophic level, e.g. dung fauna or algae, a risk is identified (PEC/PNEC ≥1) (see Introduction to Chapter 6), long-term tests for this level have to be conducted in Tier B. For the trophic levels, without an identified risk, the assessment can stop. If the risk still applies with these long-term studies, a further refinement with field studies in Tier C can be conducted. Here a co-operation with a competent authority is strongly recommended, as these tests are tailored, reflected by the individual design of these field studies. In addition, and independent of this, risk mitigation measures can be imposed to reduce the exposure concentration (PEC). These can be, beside others, that animals must remain stabled for a certain amount of time after the treatment, to ensure that the concentration of active ingredient in excreta is low enough to avoid adverse effects on dung fauna and their predators. Alternatively, the treated animals are denied access to water as the active ingredient has harmful effects on aquatic organisms.
Conclusion
The Environmental Risk Assessment of Human and Veterinary Medicinal Products is a straightforward, tiered-based process with the possibility to exit at several steps in the assessment procedure. Depending on the dose, the physico-chemical properties, and the anticipated use, this can be quite early in the procedure. On the other hand, for very potent substances with specific modes of action the guidelines are flexible enough to allow specific assessments covering these modes of action.
The ERA guideline for human medicinal products entered into application 2006 and many data gaps exist for products approved prior to 2006. Although there is a legal requirement for an ERA dossier for all marketing authorisation applications, new applications for pharmaceuticals on the market before 2006 are only required to submit ERA data under certain circumstances (e.g. significant increase in usage). Even for some of the blockbusters, like Ibuprofen, Diclofenac, and Metformin, full information on fate, behaviour and effects on non-target organisms is currently lacking.
Furthermore, systematic post-authorisation monitoring and evaluation of potential unintended ecotoxicological effects does not exist. The market authorisation for pharmaceuticals does not expire, in contrast to e.g. an authorisation of pesticides, which needs to be renewed every 10 years.
For Veterinary Medicinal Products, an in-depth ERA is necessary for food producing animals only. An ERA for non-food animals can stop with question 3 in Phase I (Figure 3) as it is considered that the use of products for companion animals leads to negligible environmental concentrations, which might not be necessarily the case. Here, the guideline does not reflect the state of the art of scientific and regulatory knowledge. For example, the market authorisation, as a pesticide or biocide, has been withdrawn or strongly restricted for some potent insecticides like imidacloprid and fipronil which both are authorised for use in companion animals.
Further Reading
Pharmaceuticals in the Environment:https://www.umweltbundesamt.de/en/publikationen/pharmaceuticals-in-the-environment-the-global
Recommendations for reducing micro-pollutants in waters: https://www.umweltbundesamt.de/publikationen/recommendations-for-reducing-micropollutants-in
Why are pharmaceuticals a problem for non-target organisms and for the environment?
How do Human pharmaceuticals enter the environment?
How do Veterinary pharmaceuticals enter the environment?
What is the general scheme of the environmental risk assessment of human and veterinary pharmaceuticals?
Why are long-term tests needed for the assessment of human pharmaceuticals, in contrast to the assessment of veterinary pharmaceuticals?
6.5.5. Water Framework Directive
under review
6.5.6. Policy on soil and groundwater regulation
Author: Frank Swartjes
Reviewers: Kees van Gestel, Ad Ragas, Dietmar Müller-Grabherr
Learning objectives:
You should be able to
- explain how different countries regulate soil contamination issues
- list some differences between different policy systems on soil and groundwater regulations
- describe how risk assessment procedures are implemented in policy
Keywords: Policy on soil contamination, Water Framework Directive, screening values comparison, Thematic Soil Strategy, Common Forum
History
As a bomb hit, soil contamination came onto the political agenda in the United States and in Europe through a number of disasters in the late 1970s and early 1980s. Starting point was the 1978 Love Canal disaster in upper New York State, USA, in which a school and a number of residences had been built on a former landfill for chemical waste disposal with thousands of tonnes of dangerous chemical wastes, and became a national media event. In Europe in 1979, the residential site of Lekkerkerk in the Netherlands became an infamous national event. Again, a residential area had been built on a former waste dump, which included chemical waste from the painting industry, and with channels and ditches that had been filled in with chemical waste-containing materials.
Since these events, soil contamination-related policies emerged one after the other in different countries in the world. Crucial elements of these policies were a benchmark date for a ban on bringing pollutants in or on the soil ('prevention'), including a strict policy, e.g. duty of care, for contaminations that are caused after the benchmark date, financial liability for polluting activities, tools for assessing the quality of soil and groundwater, and management solutions (remediation technologies and facilities for disposal).
Evolution in soil policies
Objectives in soil policies often show evolution over time and changes go along with developing new concepts and approaches for implementing policies. In general, soil policies often develop from a maximum risk control until a functional approach. The corresponding tools for implementation usually develop from a set of screening values towards a systemic use of frameworks, enabling sound environmental protection while improving the cost-benefit-balance. Consequently, soil policy implementation usually goes through different stages. In general terms, four different stages can be distinguished, i.e., related to maximum risk control, to the use of screening values, to the use of frameworks and based on a functional approach. Maximum risk control follows the precaution principle and is a stringent way of assessing and managing contamination by trying to avoid any risk. Procedures based on screening values allow for a distinction in polluted and non-polluted sites for which the former, the polluted sites, require some kind of intervention. The scientific underpinning of the earliest generations of screening values was limited and expert judgement played an important role. Later, more sophisticated screening values emerged, based on risk assessment. This resulted in screening values for individual contaminants within the contaminant groups metals and metalloids, other inorganic contaminants (e.g., cyanides), polycyclic aromatic hydrocarbons (PAHs), monocyclic aromatic hydrocarbons (including BETX (benzene, toluene, xylene)), persistent organic pollutants (including PCBs and dioxins), volatile organic contaminants (including trichloroethylene, tetrachloroethylene, 1,1,1-trichloroethane, and vinyl chloride), petroleum hydrocarbons and, in a few countries only, asbestos. For some contaminants such as PAHs, sum-screening values for groups were derived in several countries, based on toxicity equivalents. In a procedure based on frameworks, often the same screening values generally act as a trigger for further, more detailed site-specific investigations in one or two additional assessment steps. In the functional approach, soil and groundwater must be suited for the land use it relates to (e.g., agricultural or residential land) and the functions (e.g., drinking water abstraction, irrigation water) it performs. Some countries skip the maximum risk control and sometimes also the screening values stages and adopt a framework and/or a functional approach.
European collaboration and legislation
In Europe, collaboration was strengthened by concerted actions such as CARACAS (concerted action on risk assessment for contaminated sites in the European Union; 1996 - 1998) and CLARINET (Contaminated Land Rehabilitation Network for Environmental Technologies; 1998 - 2001). These concerted actions were followed up by fruitful international networks that are still are active today. These are the Common Forum, which is a network of contaminated land policy makers, regulators and technical advisors from Environment Authorities in European Union member states and European Free Trade Association countries, and NICOLE (Network for Industrially Co-ordinated Sustainable Land Management in Europe), which is a leading forum on industrially co-ordinated sustainable land management in Europe. NICOLE is promoting co-operation between industry, academia and service providers on the development and application of sustainable technologies.
In 2000, the EU Water Framework Directive (WFD; Directive 2000/60/EC) was adopted by the European Commission, followed by the Groundwater Directive (Directive 2006/118/EC) in 2006 (European parliament and the council of the European Union, 2019b). The environmental objectives are defined by the WFD. Moreover, 'good chemical status' and the 'no deterioration clause' account for groundwater bodies. 'Prevent and limit' as an objective aims to control direct or indirect contaminant inputs to groundwater, and distinguishes for 'preventing hazardous substances' to enter groundwater as well as 'limiting other non-hazardous substances'. Moreover, the European Commission adopted a Soil Thematic Strategy, with soil contamination being one out of the seven identified threats. A proposal for a Soil Framework Directive, launched in 2006, with the objective to protect soils across the EU, was formally withdrawn in 2014 because of a lack of support from some countries.
Policies in the world
Today, most countries in Europe and North America, Australia and New Zealand, and several countries in Asia and Middle and South America, have regulations on soil and groundwater contamination. The policies, however, differ substantially in stage, extent and format. Some policies only cover prevention, e.g., blocking or controlling the inputs of chemicals onto the soil surface and in groundwater bodies. Other policies cover prevention, risk based quality assessment and risk management procedures and include elaborated technical tools, which enable a decent and uniform approach. In particular the larger countries such as the USA, Germany and Spain, policies differ between states or provinces within the country. And even in countries with a policy on the federal level, the responsibilities for different steps in the soil contamination chain are very different for the different layers of authorities (at the national, regional and municipal level).
In Figure 1 the European countries are shown that have a procedure based on frameworks (as described above), including risk-based screening values. It is difficult, if not impossible, to summarise all policies on soil and groundwater protection worldwide. Alternatively, some general aspects of these policies are given here. A fair first basic element in nearly all soil and groundwater policies, relating to prevention of contamination, is the declaration of a formal point in time after which polluting soil and groundwater is considered an illegal act. For soil and groundwater quality assessment and management, most policies follow the risk-based land management approach as the ultimate form of the functional approach described above. Central in this approach are the risks for specific targets that need to be protected up to a specified level. Different protection targets are considered. Not surprisingly, 'human health' is the primary protection target that is adopted in nearly all countries with soil and groundwater regulations. Moreover, the ecosystem is an important protection target for soil, while for groundwater the ecosystem as a protection target is under discussion. Another interesting general characteristic of mature soil and groundwater policies is the function-specific approach. The basic principle of this approach is that land must be suited for its purpose. As a consequence, the appraisal of a contaminated site in a residential area, for instance, follows a much more stringent concept than that of an industrial site.

Risk assessment tools
Risk assessment tools often form the technical backbone of policies. Since the late 1980s risk assessment procedures for soil and groundwater quality appraisal were developed. In the late 1980s the exposure model CalTOX was developed by the Californian Department of Toxic Substances Control in the USA, a few years later the CSOIL model in the Netherlands (Van den Berg, 1991/ 1994/ 1995). In Figure 2, the flow chart of the Dutch CSOIL exposure model is given as an example. Three elements are recognized in CSOIL, like in most exposure models: (1) contaminant distribution over the soil compartments; (2) contaminant transfer from (the different compartments of) the soil into contact media; and (3) direct and indirect exposure to humans. The major exposure pathways are exposure through soil ingestion, crop consumption and inhalation of indoor vapours (Elert et al., 2011). Today, several exposure models exist (see Figure 3 for some 'national' European exposure models). However, these exposure models may give quite different exposure estimates for the same exposure scenario (Swartjes, 2007).


Moreover, procedures were developed for ecological risk assessment, including the Species Sensitivity Distributions (see section on SSDs), based on empirical relations between concentration in soil or groundwater and the percentage of species or ecological processes that experience adverse effects (PAF: potentially Affected Fraction). For site specific risk assessment, the TRIAD approach was developed, based on three lines of evidence, i.e., chemically-based, toxicity-based and using data from ecological field surveys (see section on the TRIAD approach).
In the framework of the HERACLES network, another attempt was made to summarizing different EU policies on polluted soil and groundwater. A strong plea was made for harmonisation of risk assessment tools (Swartjes et al., 2009). The authors also described a procedure for harmonization based on the development of a toolbox with standardized and flexible risk assessment tools. Flexible tools are meant to cover national or regional differences in cultural, climatic and geological (e.g., soil type, depth of the groundwater table) conditions. It is generally acknowledged, however, that policy decisions should be taken on the national level. In 2007, an analysis of the differences of soil and groundwater screening values and of the underlying regulatory frameworks, human health and ecological risk assessment procedures (Carlon, 2007) was launched. Although screening values are difficult to compare, since frameworks and objectives of screening values differ significantly, a general conclusion can be drawn for e.g. the screening values at the potentially unacceptable risk level (often used as 'action' values, i.e. values that trigger further research or intervention when exceeded). For the 20 metals, most soil screening values (from 13 countries or regions) show between a factor of 10 and 100 difference between the lowest and highest values. For the 23 organic pollutants considered, most soil screening values (from 15 countries or regions) differ by a factor of between 100 and 1000, but for some organic pollutants these screening values differ by more than four orders of magnitude. These conclusions are merely relevant from a policy viewpoint. Technically, these conclusions are less relevant, since, the screenings values are derived from a combination of different protection targets and tools and based on different policy decisions. Differences in screening values are explained by differences in geographical and biological and socio-cultural factors in different countries and regions, different national regulatory and policy decisions and variability in scientific/ technical tools.
References
Carlon, C. (Ed.) (2007). Derivation methods of soil screening values in Europe. A review and evaluation of national procedures towards harmonisation, JRC Scientific and Technical report EUR 22805 EN.
Elert, M., Bonnard, R., Jones, C., Schoof, R.A., Swartjes, F.A. (2011). Human Exposure Pathways. Chapter 11 in: Swartjes, F.A. (Ed.), Dealing with Contaminated Sites. From theory towards practical application. Springer Publishers, Dordrecht.
Swartjes, F.A. (2007). Insight into the variation in calculated human exposure to soil contaminants using seven different European models. Integrated Environmental Assessment and Management 3, 322-332.
Swartjes, F.A., D'Allesandro, M., Cornelis, Ch., Wcislo, E., Müller, D., Hazebrouck, B., Jones, C., Nathanail, C.P. (2009). Towards consistency in risk assessment tools for contaminated sites management in the EU. The HERACLES strategy from the end of 2009 onwards. National Institute for Public Health and the Environment (RIVM), Bilthoven, The Netherlands, RIVM Report 711701091.
Van den Berg, R. (1991/1994/1995). Exposure of humans to soil contamination. A quantitative and qualitative analyses towards proposals for human toxicological C‑quality standards (revised version of the 1991/ 1994 reports). National Institute for Public Health and the Environment (RIVM), Bilthoven, The Netherlands, RIVM-report no. 725201011.
Further reading
Swartjes, F.A. (Ed.) (2011). Dealing with Contaminated Sites. From theory towards practical application. Springer Publishers, Dordrecht.
Rodríguez-Eugenio, N., McLaughlin, M., Pennock, D. (2018). Soil Pollution: a hidden reality. Rome, FAO.
What is the logical first step in policy on soil and groundwater protection, related to ' prevention' ?
What are the most frequently used protection targets on policies on soil and groundwater in the world?
What role should screening values play in sophisticated risk assessment procedures?
What is the ideal approach when developing a new soil and groundwater policy?
Regarding the harmonization of risk assessment tools: why are flexible risk assessment tools necessary?
6.5.7. Drinking water
in preparation