
4.2: IR Spectroscopy
- Page ID
- 55873

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)IR Sample Preparation: A Practical Guide
Infrared spectroscopy is based on molecular vibrations caused by the oscillation of molecular dipoles. Bonds have characteristic vibrations depending on the atoms in the bond, the number of bonds and the orientation of those bonds with respect to the rest of the molecule. Thus, different molecules have specific spectra that can be collected for use in distinguishing products or identifying an unknown substance (to an extent.)
Collecting spectra through this method goes about one of three general ways. Nujol mulls and pressed pellets are typically used for collecting spectra of solids, while thin-film cells are used for solution-phase IR spectroscopy. Spectra of gases can also be obtained but will not be discussed in this guide.
Infrared Optical Materials and Handling
While it is all well and wonderful that substances can be characterized in this fashion one still has to be able to hold the substances inside of the instrument and properly prepare the samples. In an infrared spectrometer (Figure \(\PageIndex{1}\) )
the sample to be analyzed is held in front of an infrared laser beam, in order to do this, the sample must be contained in something, consequently this means that the very container the sample is in will absorb some of the infrared beam.

This is made somewhat complicated by the fact that all materials have some sort of vibration associated with them. Thus, if the sample holder has an optical window made of something that absorbs near where your sample does, the sample might not be distinguishable from the optical window of the sample holder. The range that is not blocked by a strong absorbance is known as a window (not to be confused with the optical materials of the cell.)
Windows are an important factor to consider when choosing the method to perform an analysis, as seen in Table \(\PageIndex{1}\) there are a number of different materials each with their own characteristic absorption spectra and chemical properties. Keep these factors in mind when performing analyses and precious sample will be saved. For most organic compounds NaCl works well though it is susceptible to attack from moisture. For metal coordination complexes KBr, or CsI typically work well due to their large windows. If money is not a problem then diamond or sapphire can be used for plates.
| Material | Transparent Ranges (cm -1) | Solubility | Notes |
|---|---|---|---|
| NaCl | 40,000 - 625 | H2O | Easy to polish, hygroscopic |
| Silica glass | 55,000-3,000 | HF | Attacked by HF |
| Quartz | 40,000-2,500 | HF | Attacked by HF |
| Sapphire | 20,000-1,780 | - | Strong |
| Diamond | 40,000-2,500 and 1,800-200 | - | Very strong, expensive, hard, useless for pellets |
| CaF2 | 70,000-1,110 | Acids | Attacked by acids, avoid ammonium salts |
| BaF2 | 65,000-700 | - | Avoid ammonium salts |
| ZnSe | 10,000 - 550 | Acids | Brittle, attacked by acids |
| AgCl | 25,000-400 | - | Soft, sensitive to light. |
| KCl | 40,000-500 | H2O, Et2O, acetone | Hygroscopic, soft, easily polished, commonly used in making pellets. |
| KBr | 40,000-400 | H2O, EtOH | Hygroscopic, soft, easily polished, commonly used in making pellets. |
| CsBr | 10,000-250 | H2O, EtOH, acetone | Hygroscopic soft |
| CsI | 10,000-200 | H2O, EtOH, MeOH, acetone | Hygroscopic, soft. |
| Teflon | 5,000-1,200; 1,200-900 | - | Inert, disposable |
| Polyethylene | 4,000-3,000; 2,800-1,460; 1,380 - 730; 720- 30 | - | Inert, disposable |
Proper handling of these plates will ensure they have a long, useful life. Here follows a few simple pointers on how to handle plates:
- Avoid contact with solvents that the plates are soluble in.
- Keep the plates in a dessicator, the less water the better, even if the plates are insoluble to water.
- Handle with gloves, clean gloves.
- Avoid wiping the plates to prevent scratching.
That said, these simple guidelines will likely reduce most damage that can occur to a plate by simply holding it other faults such as dropping the plate from a sufficient height can result in more serious damage.
Preparation of Nujol Mulls
A common method of preparing solid samples for IR analysis is mulling. The principle here is by grinding the particles to below the wavelength of incident radiation that will be passing through there should be limited scattering. To suspend those tiny particles, an oil, often referred to as Nujol is used. IR-transparent salt plates are used to hold the sample in front of the beam in order to acquire data. To prepare a sample for IR analysis using a salt plate, first decide what segment of the frequency band should be studied, refer to Table \(\PageIndex{1}\) for the materials best suited for the sample. Figure \(\PageIndex{2}\) shows the materials needed for preparing a mull.

Preparing the mull is performed by taking a small portion of sample and adding approximately 10% of the sample volume worth of the oil and grinding this in an agate mortar and pestle as demonstrated in Figure \(\PageIndex{3}\). The resulting mull should be transparent with no visible particles.

Another method involves dissolving the solid in a solvent and allowing it to dry in the agate pestle. If using this method ensure that all of the solvent has evaporated since the solvent bands will appear in the spectrum. Some gentle heating may assist this process. This method creates very fine particles that are of a relatively consistent size. After addition of the oil further mixing (or grinding) may be necessary.
Plates should be stored in a desiccator to prevent erosion by atmospheric moisture and should appear roughly transparent. Some materials such as silicon will not, however. Gently rinse the plates with hexanes to wash any residual material off of the plates. Removing the plates from the desiccator and cleaning them should follow the preparation of the mull in order to maintain the integrity of the salt plates. Of course, if the plate is not soluble in water then it is still a good idea just to prevent the threat of mechanical trauma or a stray jet of acetone from a wash bottle.
Once the mull has been prepared, add a drop to one IR plate (Figure \(\PageIndex{4}\) ), place the second plate on top of the drop and give it a quarter turn in order to evenly coat the plate surface as seen in Figure \(\PageIndex{5}\). Place it into the spectrometer and acquire the desired data.
Always handle with gloves and preferably away from any sinks, faucets, or other sources of running or spraying water.

Spectra acquired by this method will have strong C-H absorption bands throughout several ranges 3,000 – 2,800 cm-1 and 1,500 – 1,300 cm-1 and may obscure signal.
Cleaning the plate is performed as previously mentioned with hexanes or chloroform can easily be performed by rinsing and leaving them to dry in the hood. Place the salt plates back into the desiccator as soon as reasonably possible to prevent damage. It is highly advisable to polish the plates after use, no scratches, fogging, or pits should be visible on the face of the plate. Chips, so long as they don’t cross the center of the plate are survivable but not desired. The samples of damaged salt plates in Figure \(\PageIndex{6}\) show common problems associated with use or potentially mishandling. Clouding, and to an extent, scratches can be polished out with an iron rouge. Areas where the crystal lattice is disturbed below the surface are impossible to fix and chips cannot be reattached.

FIgure \(\PageIndex{6}\) A series of plates indicating various forms of physical damage with a comparison to a good plate (Copyright: Colorado University-Boulder).
Preparation of Pellets
In an alternate method, this technique is along the same lines of the nujol mull except instead of the suspending medium being mineral oil, the suspending medium is a salt. The solid is ground into a fine powder with an agate mortar and pestle with an amount of the suspending salt. Preparing pellets with diamond for the suspending agent is somewhat illadvised considering the great hardness of the substance. Generally speaking, an amount of KBr or CsI is used for this method since they are both soft salts. Two approaches can be used to prepare pellets, one is somewhat more expensive but both usually yield decent results.
The first method is the use of a press. The salt is placed into a cylindrical holder and pressed together with a ram such as the one seen in (Figure \(\PageIndex{7}\) ). Afterwards, the pellet, in the holder, is placed into the instrument and spectra acquired.

An alternate, and cheaper method requires the use of a large hex nut with a 0.5 inch inner diameter, two bolts, and two wrenches such as the kit seen in Figure \(\PageIndex{8}\). Step-by-step instructions for loading and using the press follows:
- Screw one of the bolts into the nut about half way.
- Place the salt pellet mixture into the other opening of the nut and level by tapping the assembly on a countertop.
- Screw in the second bolt and place the assembly on its side with the bolts parallel to the countertop. Place one of the wrenches on the bolt on the right side with the handle aiming towards yourself.
- Take the second wrench and place it on the other bolt so that it attaches with an angle from the table of about 45 degrees.
- The second bolt is tightened with a body weight and left to rest for several minutes. Afterwards, the bolts are removed, and the sample placed into the instrument.

Some pellet presses also have a vacuum barb such as the one seen in (Figure \(\PageIndex{8}\). If your pellet press has one of these, consider using it as it will help remove air from the salt pellet as it is pressed. This ensures a more uniform pellet and removes absorbances in the collected spectrum due to air trapped in the pellet.
Preparation of Solution Cells
Solution cells (Figure \(\PageIndex{9}\) ) are a handy way of acquiring infrared spectra of compounds in solution and is particularly handy for monitoring reactions.

A thin-film cell consists of two salt plates with a very thin space in between them (Figure \(\PageIndex{10}\) ). Two channels allow liquid to be injected and then subsequently removed. The windows on these cells can be made from a variety of IR optical materials. One particularly useful one for water-based solutions is CaF2 as it is not soluble in water.
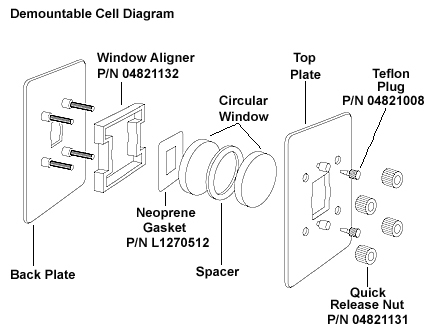
Cleaning these cells can be performed by removing the solution, flushing with fresh solvent and gently removing the solvent by syringe. Do not blow air or nitrogen through the ports as this can cause mechanical deformation in the salt window if the pressure is high enough.
Deuterated Solvent Effects
One of the other aspects to solution-phase IR is that the solvent utilized in the cell has a characteristic absorption spectra. In some cases this can be alleviated by replacing the solvent with its deuterated sibling. The benefit here is that C-H bonds are now C-D bonds and have lower vibrational frequencies. Compiled in Figure \(\PageIndex{11}\) is a set of common solvents.

This effect has numerous benefits and is often applied to determining what vibrations correspond to what bond in a given molecular sample. This is often accomplished by using isotopically labeled “heavy” reagents such as ones that contain 2H, 15N, 18O, or 13C.
Basic Troubleshooting
There are numerous problems that can arise from improperly prepared samples, this section will go through some of the common problems and how to correct them. For this demonstration, spectra of ferrocene will be used. The molecular structure and a photograph of the brightly colored organometallic compound are shown in Figure \(\PageIndex{12}\) and Figure \(\PageIndex{13}\).
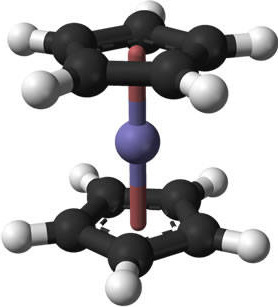

Figure \(\PageIndex{14}\) illustrates what a good sample of ferrocene looks like prepared in a KBr pellet. The peaks are well defined and sharp. No peak is flattened at 0% transmittance and Christiansen scattering is not evident in the baseline.
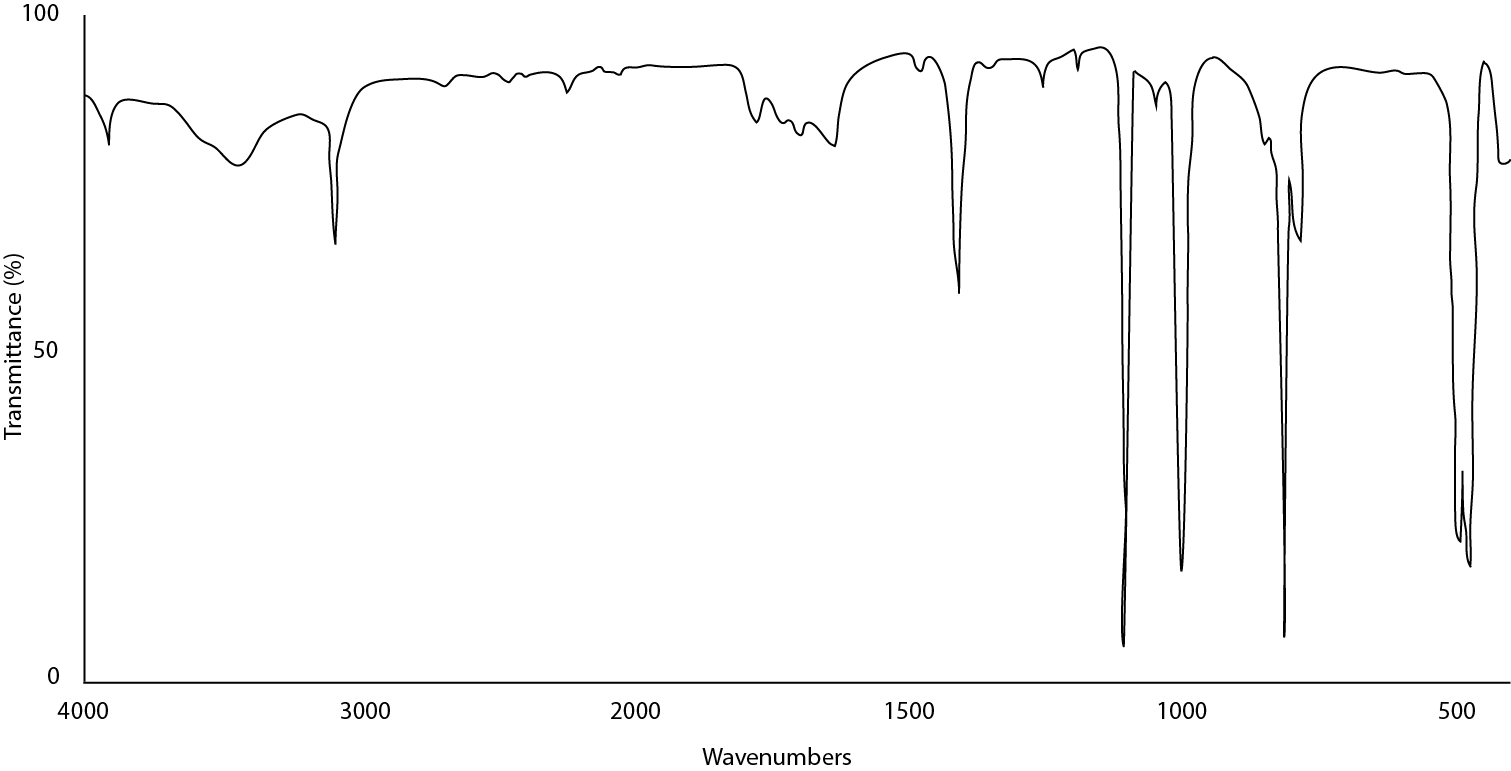
Figure \(\PageIndex{15}\) illustrates a sample with some peaks with intensities that are saturated and lose resolution making peak-picking difficult. In order to correct for this problem, scrape some of the sample off of the salt plate with a rubber spatula and reseat the opposite plate. By applying a thinner layer of sample one can improve the resolution of strongly absorbing vibrations.
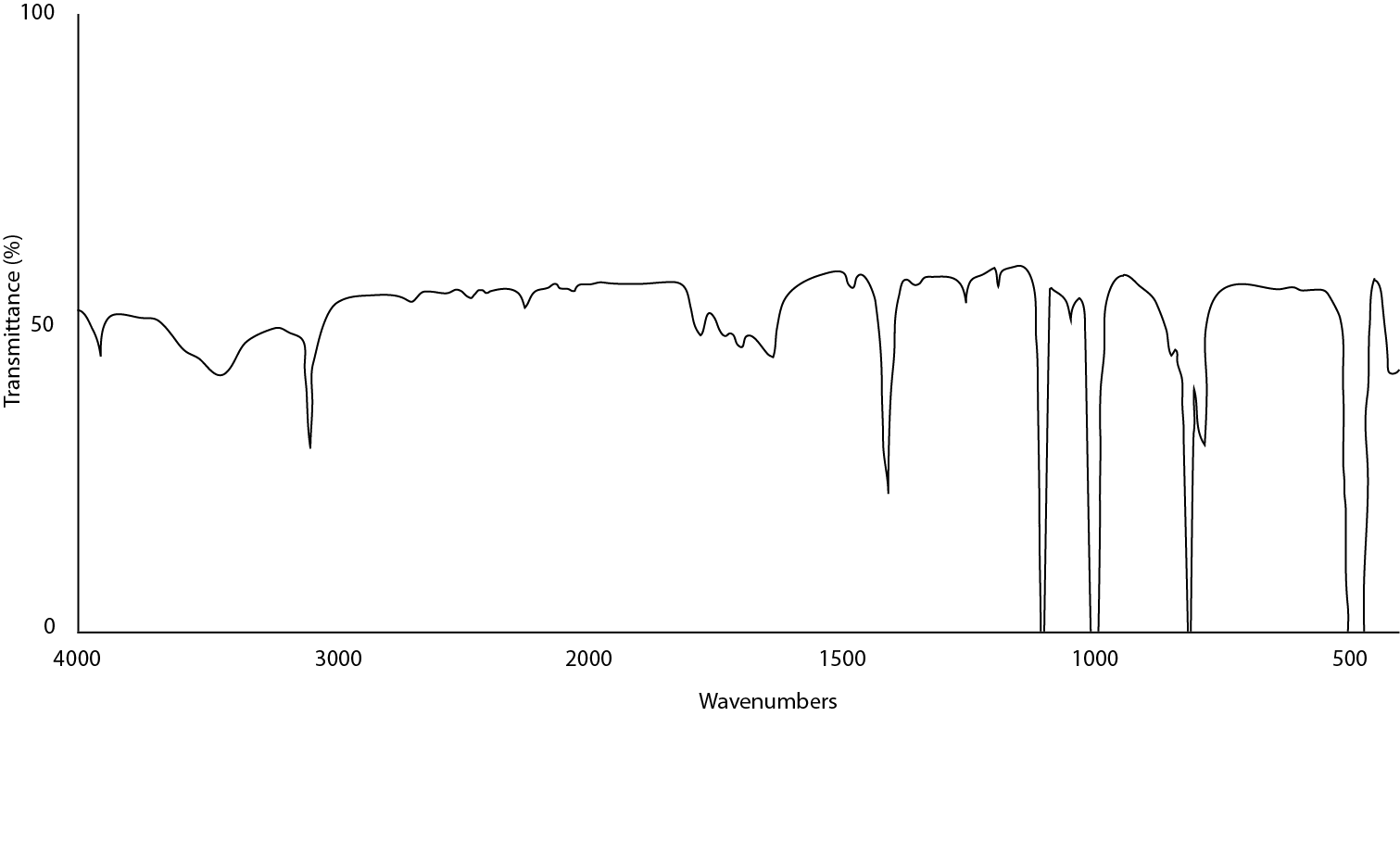
Figure \(\PageIndex{16}\) illustrates a sample in which too much mineral oil was added to the mull so that the C-H bonds are far more intense than the actual sample. This can be remedied by removing the sample from the plate, grinding more sample and adding a smaller amount of the mull to the plate. Another possible way of doing this is if the sample is insoluble in hexanes, add a little to the mull and wick away the hexane-oil mixture to leave a dry solid sample. Apply a small portion of oil and replate.
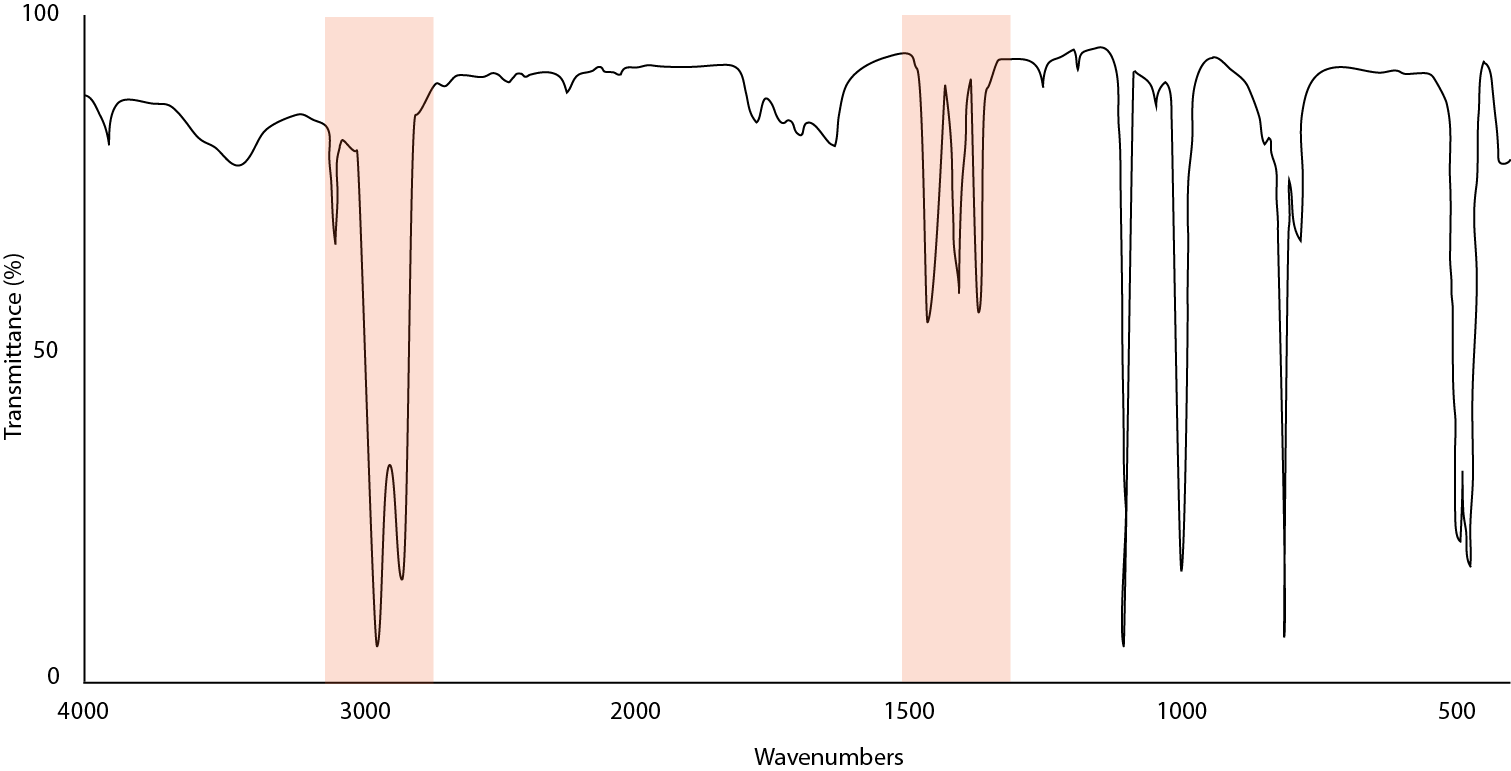
Figure \(\PageIndex{17}\) illustrates the result of particles being too large and scattering light. To remedy this, remove the mull and grind further or else use the solvent deposition technique described earlier.
Characteristic IR Vibrational Modes for Hydrocarbon Compounds
| Functional group | Mode | Wavenumber range (cm-1) |
|---|---|---|
| CH3 | Asymmetric stretch | 2962±10 |
| CH3 | Symmetric stretch | 2872±10 |
| CH3 | Asymmetric bend | 1460±10 |
| CH3 | Symmetric bend (umbrella mode) | 1375±10 |
| CH2 | Asymmetric stretch | 2926±10 |
| CH2 | Symmetric stretch | 2855±10 |
| CH2 | Scissors | 1455±10 |
| CH2 | Rock | 720±10 |
| CH | Stretch | ~2900 (weak) |
| CH | Bend | ~1350 (weak) |
| Substitution | C-H stretch (cm-1) | C=C stretch (cm-1) | Out of plane bend (cm-1) |
|---|---|---|---|
| Vinyl | 3090-3075 | 1660-1630 | 900±5, 910±5 |
| Vinylidine | 3090-3075 | 1660-1630 | 890±5 |
| Cis | 3050-3000 | 1660-1630 | 690±10 |
| Trans | 3050-3000 | 1680-1665 | 965±5 |
| Tri-substituted | 3050-3000 | 1680-1665 | 815±25 |
| Tetra-substituted | - | 1680-1665 | - |
| Substitution | C-H stretch (cm-1) | C=C stretch (cm-1) | C-H wag (cm-1) |
|---|---|---|---|
| Mono-substituted | 3350-3250 | 2140-2100 | 700-600 |
| Di-substituted | - | 2260-2190 | - |
| Substitution | Out of plane C-H bending | Ring bend (cm-1) |
|---|---|---|
| Mono | 770-710 | 690±10 |
| Ortho | 810-750 | - |
| Meta | 770-735 | 690±10 |
| Para | 860-790 | - |
| Vibration | Wavenumber (cm-1) |
|---|---|
| CH3 symmetric stretch | 2925±5 |
| CH3 bend overtone | 2865±5 |
Fourier Transform Infrared Spectroscopy of Metal Ligand Complexes
The infrared (IR) range of the electromagnetic spectrum is usually divided into three regions:
- The far-infrared is always used for rotational spectroscopy, with wavenumber range 400 – 10 cm−1 and lower energy.
- The mid-infrared is suitable for a detection of the fundamental vibrations and associated rotational-vibrational structure with the frequency range approximately 4000 – 400 cm−1.
- The near-Infrared with higher energy and wave number range 14000 – 4000 cm−1, can excite overtone or higher harmonic vibrations.
For classical light material interaction theory, if a molecule can interact with an electromagnetic field and absorb a photon of certain frequency, the transient dipole of molecular functional group must oscillate at that frequency. Correspondingly, this transition dipole moment must be a non-zero value, however, some special vibration can be IR inactive for the stretching motion of a homonuclear diatomic molecule and vibrations do not affect the molecule’s dipole moment (e.g., N2).
Mechanistic Description of the Vibrations of Polyatomic Molecules
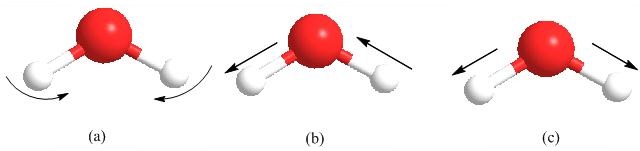
A molecule can vibrate in many ways, and each way is called a "vibrational mode". If a molecule has N atoms, linear molecules have 3N-5 degrees of vibrational modes whereas nonlinear molecules have 3N-6 degrees of vibrational modes. Take H2O for example; a single molecule of H2O has O-H bending mode (Figure \(\PageIndex{18}\) a), antisymmetric stretching mode (Figure \(\PageIndex{18}\) b), and symmetric stretching mode (Figure \(\PageIndex{18}\) c).
If a diatomic molecule has a harmonic vibration with the energy, \ref{1} , where n+1/2 with n = 0, 1, 2 ...). The motion of the atoms can be determined by the force equation, \ref{2} , where k is the force constant). The vibration frequency can be described by \ref{3} . In which m is actually the reduced mass (mred or μ), which is determined from the mass m1 and m2 of the two atoms, \ref{4} .
\[ E_{n} \ =\ -hv \label{1} \]
\[ F \ =\ -kx \label{2} \]
\[ \omega \ =\ (k/m)^{1/2} \label{3} \]
\[ m_{red} \ =\ \mu \ =\ \frac{m_{1} m_{2}}{m_{1}\ +\ m_{2} } \label{4} \]
Principle of Absorption Bands
In IR spectrum, absorption information is generally presented in the form of both wavenumber and absorption intensity or percent transmittance. The spectrum is generally showing wavenumber (cm-1) as the x-axis and absorption intensity or percent transmittance as the y-axis.
Transmittance, "T", is the ratio of radiant power transmitted by the sample (I) to the radiant power incident on the sample (I0). Absorbance (A) is the logarithm to the base 10 of the reciprocal of the transmittance (T). The absorption intensity of molecule vibration can be determined by the Lambert-Beer Law, \label{5} . In this equation, the transmittance spectra ranges from 0 to 100%, and it can provide clear contrast between intensities of strong and weak bands. Absorbance ranges from infinity to zero. The absorption of molecules can be determined by several components. In the absorption equation, ε is called molar extinction coefficient, which is related to the molecule behavior itself, mainly the transition dipole moment, c is the concentration of the sample, and l is the sample length. Line width can be determined by the interaction with surroundings.
\[ A\ =\ log(1/T) \ =\ -log(I/I_{0} )\ =\ \varepsilon c l \label{5} \]
The Infrared Spectrometer
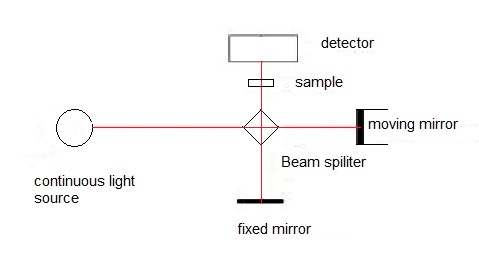
As shown in Figure \(\PageIndex{19}\), there are mainly four parts for fourier transform infrared spectrometer (FTIR):
- Light source. Infrared energy is emitted from a glowing black-body source as continuous radiations.
- Interferometer. It contains the interferometer, the beam splitter, the fixed mirror and the moving mirror. The beam splittertakes the incoming infrared beam and divides it into two optical beams. One beam reflects off the fixed mirror. The other beam reflects off of the moving mirror which moves a very short distance. After the divided beams are reflected from the two mirrors, they meet each other again at the beam splitter. Therefore, an interference pattern is generated by the changes in the relative position of the moving mirror to the fixed mirror. The resulting beam then passes through the sample and is eventually focused on the detector.
- Sample compartment. It is the place where the beam is transmitted through the sample. In the sample compartment, specific frequencies of energy are absorbed.
- Detector. The beam finally passes to the detector for final measurement. The two most popular detectors for a FTIR spectrometer are deuterated triglycine sulfate (pyroelectric detector) and mercury cadmium telluride (photon or quantum detector). The measured signal is sent to the computer where the Fourier transformation takes place.
A Typical Application: the detection of metal ligand complexes
Some General Absorption peaks for common types of functional groups
It is well known that all molecules chemicals have distinct absorption regions in the IR spectrum. Table \(\PageIndex{7}\) shows the absorption frequencies of common types of functional groups. For systematic evaluation, the IR spectrum is commonly divided into some sub-regions.
- In the region of 4000 - 2000 cm–1, the appearance of absorption bands usually comes from stretching vibrations between hydrogen and other atoms. The O-H and N-H stretching frequencies range from 3700 - 3000 cm–1. If hydrogen bond forms between O-H and other group, it generally caused peak line shape broadening and shifting to lower frequencies. The C-H stretching bands occur in the region of 3300 - 2800 cm–1. The acetylenic C-H exhibits strong absorption at around 3300 cm–1. Alkene and aromatic C-H stretch vibrations absorb at 3200-3000 cm–1. Generally, asymmetric vibrational stretch frequency of alkene C-H is around 3150 cm-1, and symmetric vibrational stretch frequency is between 3100 cm-1 and 3000 cm-1. The saturated aliphatic C-H stretching bands range from 3000 - 2850 cm–1, with absorption intensities that are proportional to the number of C-H bonds. Aldehydes often show two sharp C-H stretching absorption bands at 2900 - 2700 cm–1. However, in water solution, C-H vibrational stretch is much lower than in non-polar solution. It means that the strong polarity solution can greatly reduce the transition dipole moment of C-H vibration.
- Furthermore, the stretching vibrations frequencies between hydrogen and other heteroatoms are between 2600 - 2000cm-1, for example, S-H at 2600 - 2550 cm–1, P-H at 2440 - 2275 cm–1, Si-H at 2250 - 2100 cm–1.
- The absorption bands at the 2300 - 1850 cm–1 region usually present only from triple bonds, such as C≡C at 2260 - 2100 cm–1, C≡N at 2260 - 2000 cm–1, diazonium salts –N≡N at approximately 2260 cm–1, allenes C=C=C at 2000 - 1900 cm–1. The peaks of these groups are all have strong absorption intensities. The 1950 - 1450 cm–1 region stands for double-bonded functional groups vibrational stretching.
- Most carbonyl C=O stretching bands range from 1870 - 1550 cm–1, and the peak intensities are from mean to strong. Conjugation, ring size, hydrogen bonding, and steric and electronic effects can lead to significant shifts in absorption frequencies. Furthermore, if carbonyl links with electron-withdrawing group, such as acid chlorides and acid anhydrides, it would give rise to IR bands at 1850 - 1750 cm–1. Ketones usually display stretching bands at 1715 cm-1.
- None conjugated aliphatic C=C and C=N have absorption bands at 1690 - 1620 cm–1. Besides, around 1430 and 1370cm-1, there are two identical peaks presenting C-H bending.
- The region from 1300 - 910 cm–1 always includes the contributions from skeleton C-O and C-C vibrational stretches, giving additional molecular structural information correlated with higher frequency areas. For example, ethyl acetate not only shows its carbonyl stretch at 1750 - 1735 cm–1, but also exhibits its identical absorption peaks at 1300 - 1000 cm–1 from the skeleton vibration of C-O and C-C stretches.
| Group | Frequency (cm-1) | Strength Appearance |
| C-H stretch | 2850-3400 | Strong in nonpolar solvent Weak in polar solvent |
| O-H stretch, N-H stretch | 3200-3700 | Broad in solvent |
| C≡N stretch, R-N=C=S stretch |
2050-2300 | Medium or strong |
| C≡O stretch (bound with metal) | around 2000 | Medium or strong |
| C≡C stretch | 2100-2260 | Weak |
| C=O stretch | ca 1715 (ketone), ca 1650 (amides) |
Strong |
| C=C stretch | 1450-1700 | Weak to strong |
| C-H bend | 1260 - 1470 | Strong |
| C-O stretch | 1040-1300 | Medium or strong |
General Introduction of Metal Ligand Complex
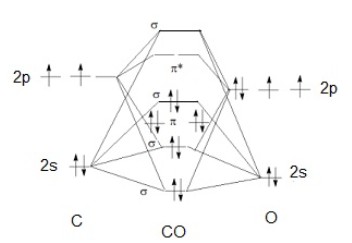
The metal electrons fill into the molecular orbital of ligands (CN, CO, etc.) to form complex compound. As shown in Figure \(\PageIndex{20}\), a simple molecular orbital diagram for CO can be used to explain the binding mechanism.
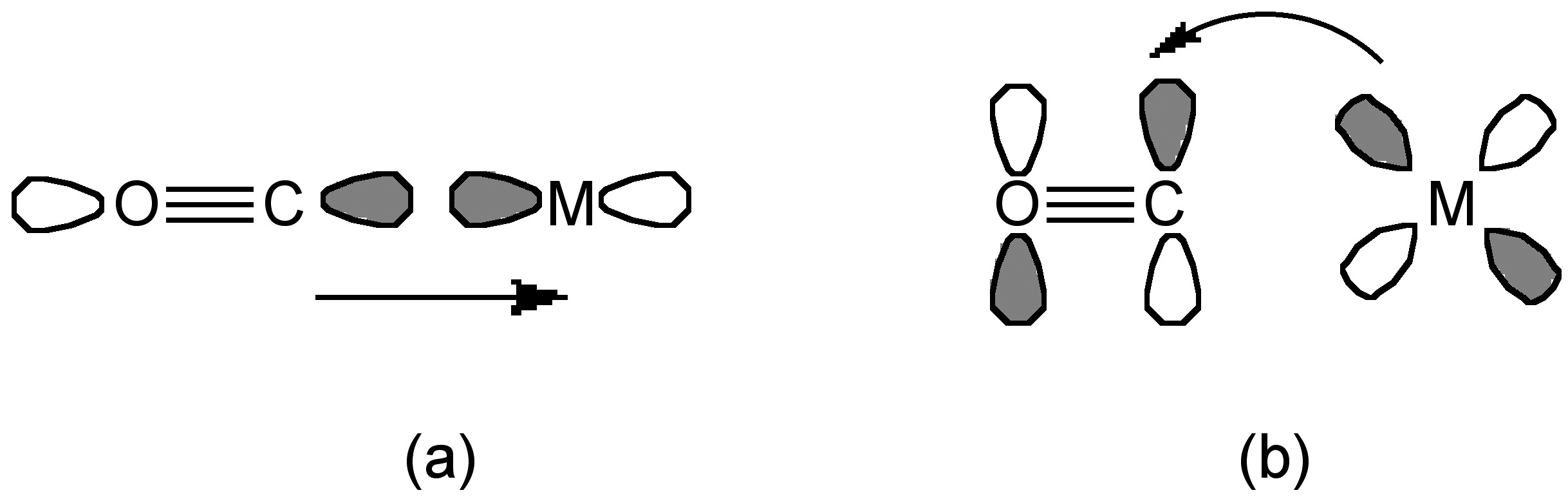
The CO and metal can bind with three ways:
- Donation of a pair of electrons from the C-O σ* orbital into an empty metal orbital (Figure \(\PageIndex{21}\) a).
- Donation from a metal d orbital into the C-O π* orbital to form a M-to-CO π-back bond (Figure \(\PageIndex{21}\) b).
- Under some conditions a pair of carbon π electron can donate into an empty metal d-orbital.
Some Factors to Include the Band Shifts and Strength
Herein, we mainly consider two properties: ligand stretch frequency and their absorption intensity. Take the ligand CO for example again. The frequency shift of the carbonyl peaks in the IR mainly depends on the bonding mode of the CO (terminal or bridging) and electron density on the metal. The intensity and peak numbers of the carbonyl bands depends on some factors: CO ligands numbers, geometry of the metal ligand complex and fermi resonance.
Effect on Electron Density on Metal
As shown in Table \(\PageIndex{8}\), a greater charge on the metal center result in the CO stretches vibration frequency decreasing. For example, [Ag(CO)]+show higher frequency of CO than free CO, which indicates a strengthening o
f the CO bond. σ donation removes electron density from the nonbonding HOMO of CO. From Figure, it is clear that the HOMO has a small amount of anti-bonding property, so removal of an electron actually increases (slightly) the CO bond strength. Therefore, the effect of charge and electronegativity depends on the amount of metal to CO π-back bonding and the CO IR stretching frequency.
| dx | Complex | CO stretch frequency (cm-1) |
| free CO | 2143 | |
| d10 | [Ag(CO)]+ | 2204 |
| d10 | Ni(CO)4 | 2060 |
| d10 | [Co(CO)4]- | 1890 |
| d6 | [Mn(CO)6]+ | 2090 |
| d6 | Cr(CO)6 | 2000 |
| d6 | [V(CO)6]- | 1860 |
If the electron density on a metal center is increasing, more π-back bonding to the CO ligand(s) will also increase, as shown in Table \(\PageIndex{9}\). It means more electron density would enter into the empty carbonyl π* orbital and weaken the C-O bond. Therefore, it makes the M-CO bond strength increasing and more double-bond-like (M=C=O).
Ligation Donation Effect
Some cases, as shown in Table \(\PageIndex{9}\), different ligands would bind with same metal at the same metal-ligand complex. For example, if different electron density groups bind with Mo(CO)3 as the same form, as shown in Figure \(\PageIndex{22}\), the CO vibrational frequencies would depend on the ligand donation effect. Compared with the PPh3 group, CO stretching frequency which the complex binds the PF3 group (2090, 2055 cm-1) is higher. It indicates that the absolute amount of electron density on that metal may have certain effect on the ability of the ligands on a metal to donate electron density to the metal center. Hence, it may be explained by the Ligand donation effect. Ligands that are trans to a carbonyl can have a large effect on the ability of the CO ligand to effectively π-backbond to the metal. For example, two trans π-backbonding ligands will partially compete for the same d-orbital electron density, weakening each other’s net M-L π-backbonding. If the trans ligand is a π-donating ligand, the free metal to CO π-backbonding can increase the M-CO bond strength (more M=C=O character). It is well known that pyridine and amines are not those strong π-donors. However, they are even worse π-backbonding ligands. So the CO is actually easy for π-back donation without any competition. Therefore, it naturally reduces the CO IR stretching frequencies in metal carbonyl complexes for the ligand donation effect.
| Metal Ligand Complex | CO Stretch Frequency (cm-1) |
|---|---|
| Mo(CO)3(PF3)3 | 2090, 2055 |
| Mo(CO)3[P(OMe)3]3 | 1977, 1888 |
| Mo(CO)3(PPh3)3 | 1934, 1835 |
| Mo(CO)3(NCCH3)3 | 1915, 1783 |
| Mo(CO)3(pyridine)3 | 1888, 1746 |

Geometry Effects
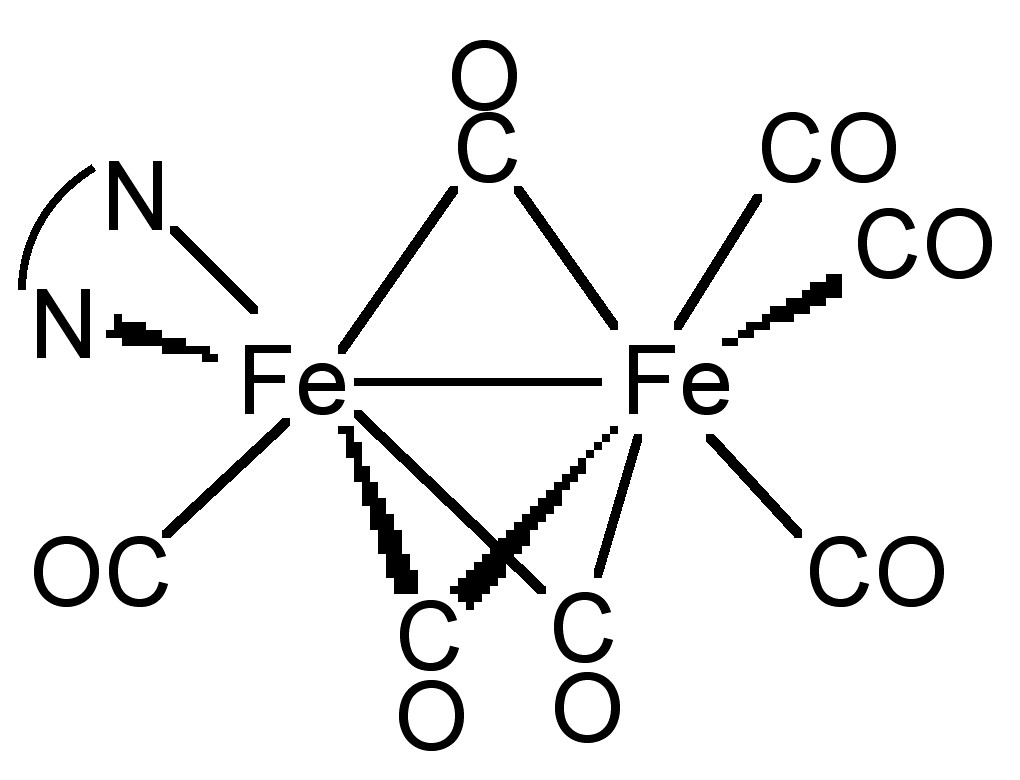
Some cases, metal-ligand complex can form not only terminal but also bridging geometry. As shown in Figure \(\PageIndex{23}\), in the compound Fe2(CO)7(dipy), CO can act as a bridging ligand. Evidence for a bridging mode of coordination can be easily obtained through IR spectroscopy. All the metal atoms bridged by a carbonyl can donate electron density into the π* orbital of the CO and weaken the CO bond, lowering vibration frequency of CO. In this example, the CO frequency in terminal is around 2080 cm-1, and in bridge, it shifts to around 1850 cm-1.
Pump-probe Detection of Molecular Functional Group Vibrational Lifetime
The dynamics of molecular functional group plays an important role during a chemical process, chemical bond forming and breaking, energy transfer and other dynamics happens within picoseconds domain. It is very difficult to study such fast processes directly, for decades scientists can only learn from theoretical calculations, lacking experimental methods.
However, with the development of ultrashort pulsed laser enable experimental study of molecular functional group dynamics. With ultrafast laser technologies, people develop a series of measuring methods, among which, pump-probe technique is widely used to study the molecular functional group dynamics. Here we concentrate on how to use pump-probe experiment to measure functional group vibrational lifetime. The principle, experimental setup and data analysis will be introduced.
Principles of the Pump-probe Technique
For every function group within a molecule, such as the C≡N triple bond in phenyl selenocyanate (C6H5SeCN) or the C-D single bond in deuterated chloroform (DCCl3), they have an individual infrared vibrational mode and associated energy levels. For a typical 3-level system (Figure \(\PageIndex{24}\), both the 0 to 1 and the 1 to 2 transition are near the probe pulse frequency (they don't necessarily need to have exactly the same frequency).
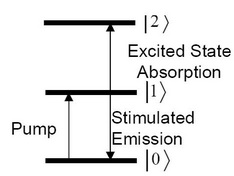
In a pump-probe experiment, we use the geometry as is shown in Figure \(\PageIndex{25}\). Two synchronized laser beams, one of which is called pump beam (Epu) while the other probe beam (Epr). There is a delay in time between each pulse. The laser pulses hit the sample, the intensity of ultrafast laser (fs or ps) is strong enough to generated 3rd order polarization and produce 3rd order optical response signal which is use to give dynamics information of molecular function groups. For the total response signals we have \label{6} , where µ10 µ21 are transition dipole moment and E0, E1, and E2 are the energies of the three levels, and t3 is the time delay between pump and probe beam. The delay t3 is varied and the response signal intensity is measured. The functional group vibration life time is determined from the data.

\[ S \ =\ 4 \mu _{10} ^{4} e^{ -i(E_{1} - E_{0} ) t3/h - \Gamma t3} \label{6} \]
Typical Experimental Set-up
The optical layout of a typical pump-probe setup is schematically displayed in Figure \(\PageIndex{26}\). In the setup, the output of the oscillator (500 mW at 77 MHz repetition rate, 40 nm bandwidth centered at 800 nm) is split into two beams (1:4 power ratio). Of this, 20% of the power is to seed a femtosecond (fs) amplifier whose output is 40 fs pulses centered at 800 nm with power of ~3.4 W at 1 KHz repetition rate. The rest (80%) of the seed goes through a bandpass filter centered at 797.5nm with a width of 0.40 nm to seed a picosecond (ps) amplifier. The power of the stretched seed before entering the ps amplifier cavity is only ~3 mW. The output of the ps amplifier is 1ps pulses centered at 800 nm with a bandwidth ~0.6 nm. The power of the ps amplifier output is ~3 W. The fs amplifier is then to pump an optical parametric amplifier (OPA) which produces ~100 fs IR pulses with bandwidth of ~200 cm-1 that is tunable from 900 to 4000 cm-1. The power of the fs IR pulses is 7~40 mW, depending on the frequencies. The ps amplifier is to pump a ps OPA which produces ~900 fs IR pulses with bandwidth of ~21 cm-1, tunable from 900 - 4000 cm-1. The power of the fs IR pulses is 10 ~ 40 mW, depending on frequencies.
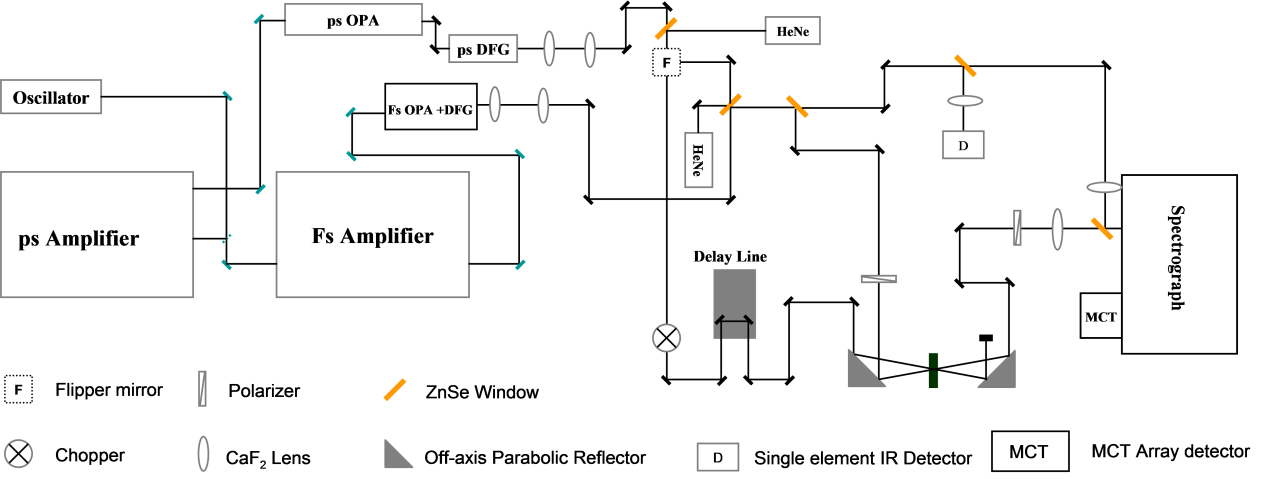
In a typical pump-probe setup, the ps IR beam is collimated and used as the pump beam. Approximately 1% of the fs IR OPA output is used as the probe beam whose intensity is further modified by a polarizer placed before the sample. Another polarizer is placed after the sample and before the spectrograph to select different polarizations of the signal. The signal is then sent into a spectrograph to resolve frequency, and detected with a mercury cadmium telluride (MCT) dual array detector. Use of a pump pulse (femtosecond, wide band) and a probe pulse (picoseconds, narrow band), scanning the delay time and reading the data from the spectrometer, will give the lifetime of the functional group. The wide band pump and spectrometer described here is for collecting multiple group of pump-probe combination.
Data Analysis
For a typical pump-probe curve shown in Figure \(\PageIndex{27}\) life time t is defined as the corresponding time value to the half intensity as time zero.
Table \(\PageIndex{10}\) shows the pump-probe data of the C≡N triple bond in a series of aromatic cyano compounds: n-propyl cyanide (C3H7CN), ethyl thiocyanate (C2H5SCN), and ethyl selenocyanate (C2H5SeCN) for which the νC≡N for each compound (measured in CCl4 solution) is 2252 cm-1), 2156 cm-1, and ~2155 cm-1, respectively.
| Delay (ps) | C3H7CN | C2H5SCN | C2H5SeCN |
|---|---|---|---|
| 0 | -0.00695 | -0.10918 | -0.06901 |
| 0.1 | -0.0074 | -0.10797 | -0.07093 |
| 0.2 | -0.00761 | -0.1071 | -0.07247 |
| 0.3 | -0.00768 | -0.10545 | -0.07346 |
| 0.4 | -0.0076 | -0.10487 | -0.07429 |
| 0.5 | -0.00778 | -0.10287 | -0.07282 |
| 0.6 | -0.00782 | -0.10286 | -0.07235 |
| 0.7 | -0.00803 | -0.10222 | -0.07089 |
| 0.8 | -0.00764 | -0.10182 | -0.07073 |
| 0.9 | -0.00776 | -0.10143 | -0.06861 |
| 1 | -0.00781 | -0.10099 | -0.06867 |
| 1.1 | -0.00745 | -0.10013 | -0.06796 |
| 1.2 | -0.00702 | -0.10066 | -0.06773 |
| 1.3 | -0.00703 | -0.0989 | -0.0676 |
| 1.4 | -0.00676 | -0.0995 | -0.06638 |
| 1.5 | -0.00681 | -0.09757 | -0.06691 |
| 1.6 | -0.00639 | -0.09758 | -0.06696 |
| 1.7 | -0.00644 | -0.09717 | -0.06583 |
| 1.8 | -0.00619 | -0.09741 | -0.06598 |
| 1.9 | -0.00613 | -0.09723 | -0.06507 |
| 2 | -0.0066 | -0.0962 | -0.06477 |
| 2.5 | -0.00574 | -0.09546 | -0.0639 |
| 3 | -0.0052 | -0.09453 | -0.06382 |
| 3.5 | -0.0482 | -0.09353 | -0.06389 |
| 4 | -0.0042 | -0.09294 | -0.06287 |
| 4.5 | -0.00387 | -0.09224 | -0.06197 |
| 5 | -0.00351 | -0.09009 | -0.06189 |
| 5.5 | -0.00362 | -0.09084 | -0.06188 |
| 6 | -0.00352 | -0.08938 | -0.06021 |
| 6.5 | -0.00269 | -0.08843 | -0.06028 |
| 7 | -0.00225 | -0.08788 | -0.05961 |
| 7.5 | -0.00231 | -0.08694 | -0.06065 |
| 8 | -0.00206 | -0.08598 | -0.05963 |
| 8.5 | -0.00233 | -0.08552 | -0.05993 |
| 9 | -0.00177 | -0.08503 | -0.05902 |
| 9.5 | -0.00186 | -0.08508 | -0.05878 |
| 10 | -0.00167 | -0.0842 | -0.0591 |
| 11 | -0.00143 | -0.08295 | -0.05734 |
A plot of intensity versus time for the data from TABLE is shown Figure \(\PageIndex{28}\). From these curves the C≡N stretch lifetimes can be determined for C3H7CN, C2H5SCN, and C2H5SeCN as ~5.5 ps, ~84 ps, and ~282 ps, respectively.
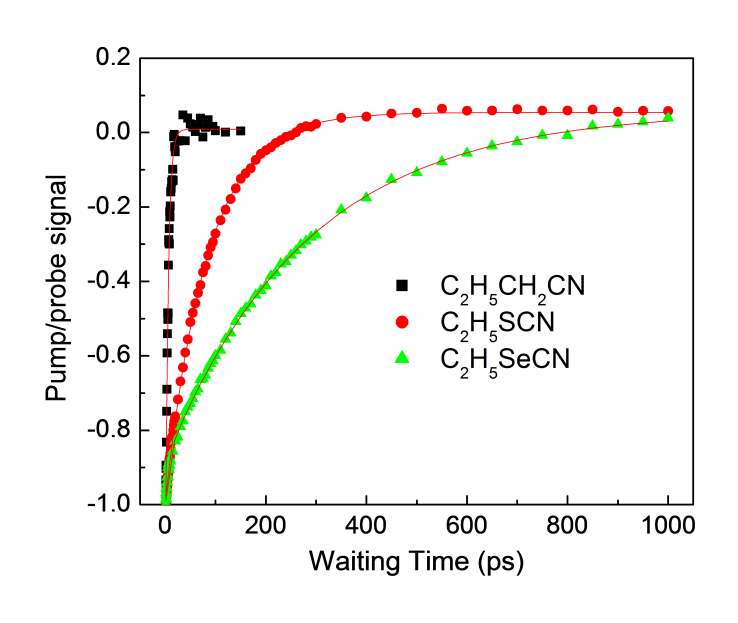
From what is shown above, the pump-probe method is used in detecting C≡N vibrational lifetimes in different chemicals. One measurement only takes several second to get all the data and the lifetime, showing that pump-probe method is a powerful way to measure functional group vibrational lifetime.
Attenuated Total Reflectace- Fourier Transform Infrared Spectroscopy
Attenuated total reflectance-Fourier transform infrared spectroscopy (ATR-FTIR) is a physical method of compositional analysis that builds upon traditional transmission FTIR spectroscopy to minimize sample preparation and optimize reproducibility. Condensed phase samples of relatively low refractive index are placed in close contact with a crystal of high refractive index and the infrared (IR) absorption spectrum of the sample can be collected. Based on total internal reflection, the absorption spectra of ATR resemble those of transmission FTIR. To learn more about transmission IR spectroscopy (FTIR) please refer to the section further up this page titled Fourier Transform Infrared Spectroscopy of Metal Ligand Complexes.
First publicly proposed in 1959 by Jacques Fahrenfort from the Royal Dutch Shell laboratories in Amsterdam, ATR IR spectroscopy was described as a technique to effectively measure weakly absorbing condensed phase materials. In Fahrenfort's first article describing the technique, published in 1961, he used a hemicylindrical ATR crystal (see Experimental Conditions) to produce single-reflection ATR (Figure \(\PageIndex{29}\) ). ATR IR spectroscopy was slow to become accepted as a method of characterization due to concerns about its quantitative effectiveness and reproducibility. The main concern being the sample and ATR crystal contact necessary to achieve decent spectral contrast. In the late 1980’s FTIR spectrometers began improving due to an increased dynamic range, signal to noise ratio, and faster computers. As a result ATR-FTIR also started gaining traction as an efficient spectroscopic technique. These days ATR accessories are often manufactured to work in conjunction with most FTIR spectrometers, as can be seen in Figure \(\PageIndex{30}\).


Total Internal Reflection
For additional information on light waves and their properties please refer to the module on Vertical Scanning Interferometry (VSI) in chapter 10.1.
When considering light propagating across an interface between two materials with different indices of refraction, the angle of refraction can be given by Snell’s law, \ref{7} , where none of the incident light will be transmitted.
\[ \varphi _{c} \ =\ \varphi _{max} \label{7} \]
The reflectance of the interface is total and whenever light is incident from a higher refractive index medium onto a lower refractive index medium, the reflection is deemed internal (as opposed to external in the opposite scenario). Total internal reflectance experiences no losses, or no transmitted light (Figure \(\PageIndex{31}\)
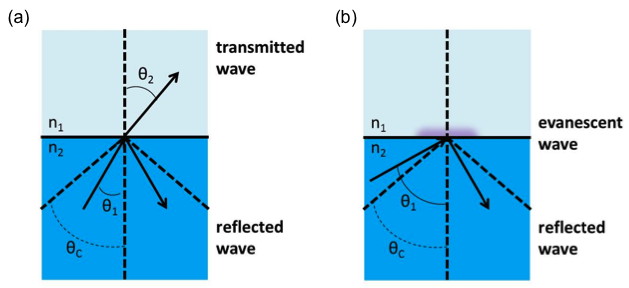
Supercritical internal reflection refers to angles of incidence above the critical angle of incidence allowing total internal reflectance. It is in this angular regime where only incident and reflected waves will be present. The transmitted wave is confined to the interface where its amplitude is at a maximum and will damp exponentially into the lower refractive index medium as a function of distance. This wave is referred to as the evanescent wave and it extends only a very short distance beyond the interface.
To apply total internal reflection to the experimental setup in ATR, consider n2 to be the internal reflectance element or ATR crystal (the blue trapezoid in Figure \(\PageIndex{32}\) )
where n2 is the material with the higher index of refraction. This should be a material that is fully transparent to the incident infrared radiation to give a real value for the refractive index. The ATR crystal must also have a high index of refraction to allow total internal reflection with many samples that have an index of refraction n1, where n1<n2.
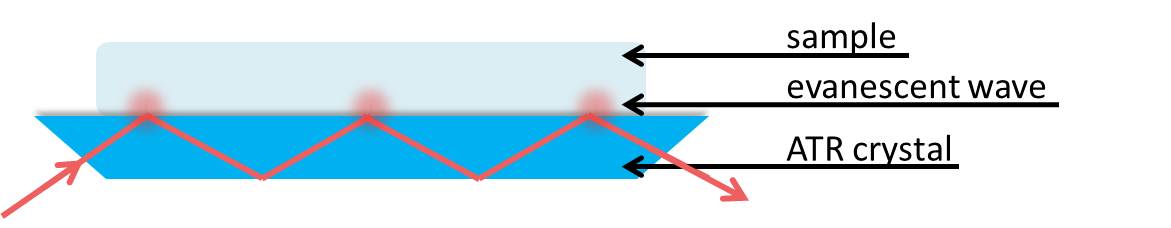
We can consider the sample to be absorbing in the infrared. Electromagnetic energy will pass through the crystal/sample interface and propagate into the sample via the evanescent wave. This energy loss must be compensated with the incident IR light. Thus, total reflectance is no longer occurring and the reflection inside the crystal is attenuated. If a sample does not absorb, the reflectance at the interface shows no attenuation. Therefore if the IR light at a particular frequency does not reach the detector, the sample must have absorbed it.
The penetration depth of the evanescent wave within the sample is on the order of 1µm. The expression of the penetration depth is given in \ref{8} and is dependent upon the wavelength and angle of incident light as well as the refractive indices of the ATR crystal and sample. The effective path length is the product of the depth of penetration of the evanescent wave and the number of points that the IR light reflects at the interface between the crystal and sample. This path length is equivalent to the path length of a sample in a traditional transmission FTIR setup.
\[ d_{p} = \frac{ \lambda }{2 \pi n_{1}} (sin \omega - ( \frac{n_{1}}{n_{2}} )^{2} )^{1/2} \label{8} \]
Experimental Conditions
Refractive Indices of ATR Crystal and Sample
Typically an ATR attachment can be used with a traditional FTIR where the beam of incident IR light enters a horizontally positioned crystal with a high refractive index in the range of 1.5 to 4, as can be seen in Table \(\PageIndex{11}\) will consist of organic compounds, inorganic compounds, and polymers which have refractive indices below 2 and can readily be found on a database.
| Material | Refractive Index (RI) | Spectral Range (cm-1) |
|---|---|---|
| Zinc Selenide (ZnSe) | 2.4 | 20,000 - 650 |
| Germanium (Ge) | 4 | 5,500 - 870 |
| Sapphire (Al2O3) | 1.74 | 50,000 - 2,000 |
| Diamond (C) | 2.4 | 45,000 - 2,500, 1650 - 200 |
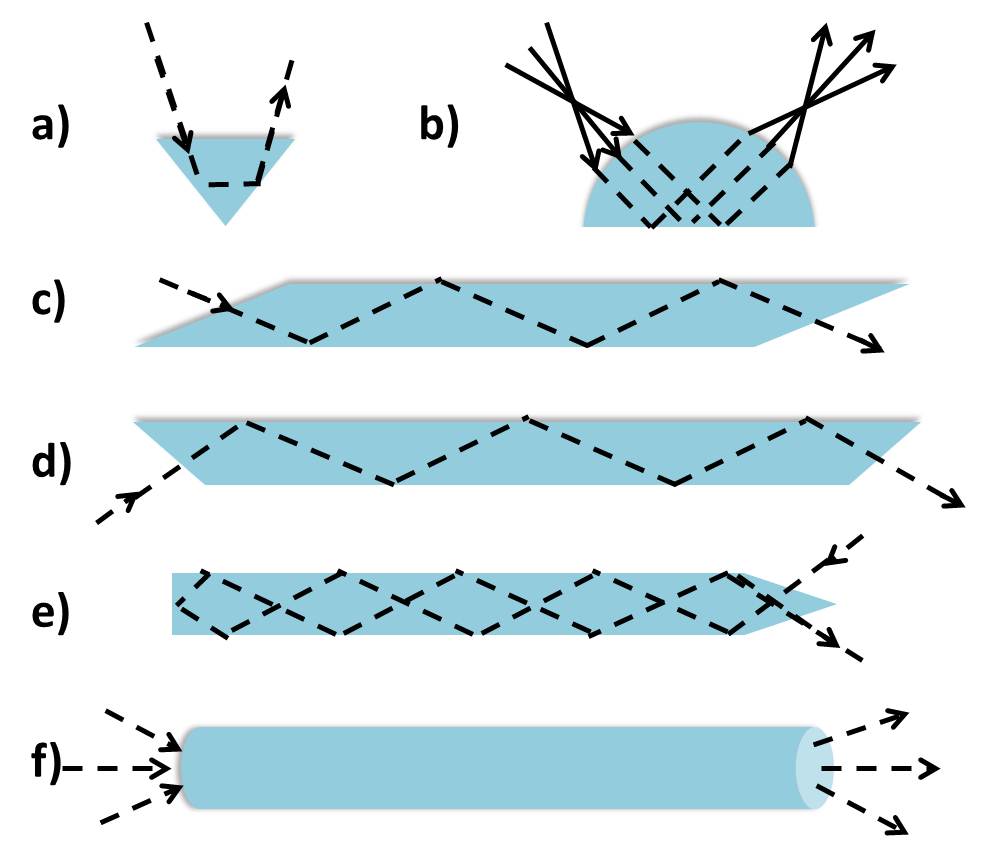
Single and Multiple Reflection Crystals
Multiple reflection ATR was initially more popular than single reflection ATR because of the weak absorbances associated with single reflection ATR. More reflections increased the evanescent wave interaction with the sample, which was believed to increase the signal to noise ratio of the spectrum. When IR spectrometers developed better spectral contrast, single reflection ATR became more popular. The number of reflections and spectral contrast increases with the length of the crystal and decreases with the angle of incidence as well as thickness. Within multiple reflection crystals some of the light is transmitted and some is reflected as the light exits the crystal, resulting in some of the light going back through the crystal for a round trip. Therefore, light exiting the ATR crystal contains components that experienced different number of reflections at the crystal-sample interface.
Angle of Incidence
It was more common in earlier instruments to allow selection of the incident angle, sometimes offering selection between 30°, 45°, and 60°. In all cases for total internal reflection to hold, the angle of incidence must exceed the critical angle and ideally complement the angle of the crystal edge so that the light enters at a normal angle of incidence. These days 45° is the standard angle on most ATR-FTIR setups.
ATR Crystal Shape
For the most part ATR crystals will have a trapezoidal shape as shown in Figure \(\PageIndex{31}\). This shape facilitates sample preparation and handling on the crystal surface by enabling the optical setup to be placed below the crystal. However, different crystal shapes (Figure \(\PageIndex{33}\) ) may be used for particular purposes, whether it is to achieve multiple reflections or reduce the spot size. For example, a hemispherical crystal may be used in a microsampling experiment in which the beam diameter can be reduced at no expense to the light intensity. This allows appropriate measurement of a small sample without compromising the quality of the resulting spectral features.
Crystal-sample contact
Because the path length of the evanescent wave is confined to the interface between the ATR crystal and sample, the sample should make firm contact with the ATR crystal (Figure \(\PageIndex{34}\) ). The sample sits atop the crystal and intimate contact can be ensured by applying pressure above the sample. However, one must be mindful of the ATR crystal hardness. Too much pressure may distort the crystal and affect the reproducibility of the resulting spectrum.

The wavelength effect expressed in \label{7} shows an increase in penetration depth at increased wavelength. In terms of wavenumbers the relationship becomes inverse. At 4000 cm-1 penetration of the sample is 10x less than penetration at 400 cm-1 meaning the intensity of the peaks may appear higher at lower wavenumbers in the absorbance spectrum compared to the spectral features in a transmission FTIR spectrum (if an automated correction to the ATR setup is not already in place).
Selecting an ATR Crystal
ATR functions effectively on the condition that the refractive index of the crystal is of a higher refractive index than the sample. Several crystals are available for use and it is important to select an appropriate option for any given experiment (Table \(\PageIndex{11}\) ).
When selecting a material, it is important to consider reactivity, temperature, toxicity, solubility, and hardness.
The first ATR crystals in use were KRS-5, a mixture of thallium bromide and iodide, and silver halides. These materials are not listed in the table because they are not in use any longer. While cost-effective, they are not practical due to their light sensitivity, softness, and relatively low refractive indices. In addition KRS-5 is terribly toxic and dissolves on contact with many solvents, including water.
At present diamond is a favorable option for its hardness, inertness and wide spectral range, but may not be a financially viable option for some experiments. ZnSe and germanium are the most common crystal materials. ZnSe is reasonably priced, has significant mechanical strength and a long endurance. However, the surface will become etched with exposure to chemicals on either extreme of the pH scale. With a strong acid ZnSe will react to form toxic hydrogen selenide gas. ZnSe is also prone to oxidation and care must be taken to avoid the formation of an IR absorbing layer of SeO2. Germanium has a higher refractive index, which reduces the depth of penetration to 1 µm and may be preferable to ZnSe in applications involving intense sample absorptions or for use with samples that produce strong background absorptions. Sapphire is physically robust with a wide spectral range, but has a relatively low refractive index in terms of ATR crystals, meaning it may not be able to test as many samples as another crystal might.
Sample Versatility
Solids
The versatility of ATR is reflected in the various forms and phases that a sample can assume. Solid samples need not be compressed into a pellet, dispersed into a mull or dissolve in a solution. A ground solid sample is simply pressed to the surface of the ATR crystal. For hard samples that may present a challenge to grind into a fine solid, the total area in contact with the crystal may be compromised unless small ATR crystals with exceptional durability are used (e.g., 2 mm diamond). Loss of contact with the crystal would result in decreased signal intensity because the evanescent wave may not penetrate the sample effectively. The inherently short path length of ATR due to the short penetration depth (0.5-5 µm) enables surface-modified solid samples to be readily characterized with ATR.
Powdered samples are often tedious to prepare for analysis with transmission spectroscopy because they typically require being made into a KBr pellet to and ensuring the powdered sample is ground up sufficiently to reduce scattering. However, powdered samples require no sample preparation when taking the ATR spectra. This is advantageous in terms of time and effort, but also means the sample can easily be recovered after analysis.
Liquids
The advantage of using ATR to analyze liquid samples becomes apparent when short effective path lengths are required. The spectral reproducibility of liquid samples is certain as long as the entire length of the crystal is in contact with the liquid sample, ensuring the evanescent wave is interacting with the sample at the points of reflection, and the thickness of the liquid sample exceeds the penetration depth. A small path length may be necessary for aqueous solutions in order to reduce the absorbance of water.
Sample Preparation
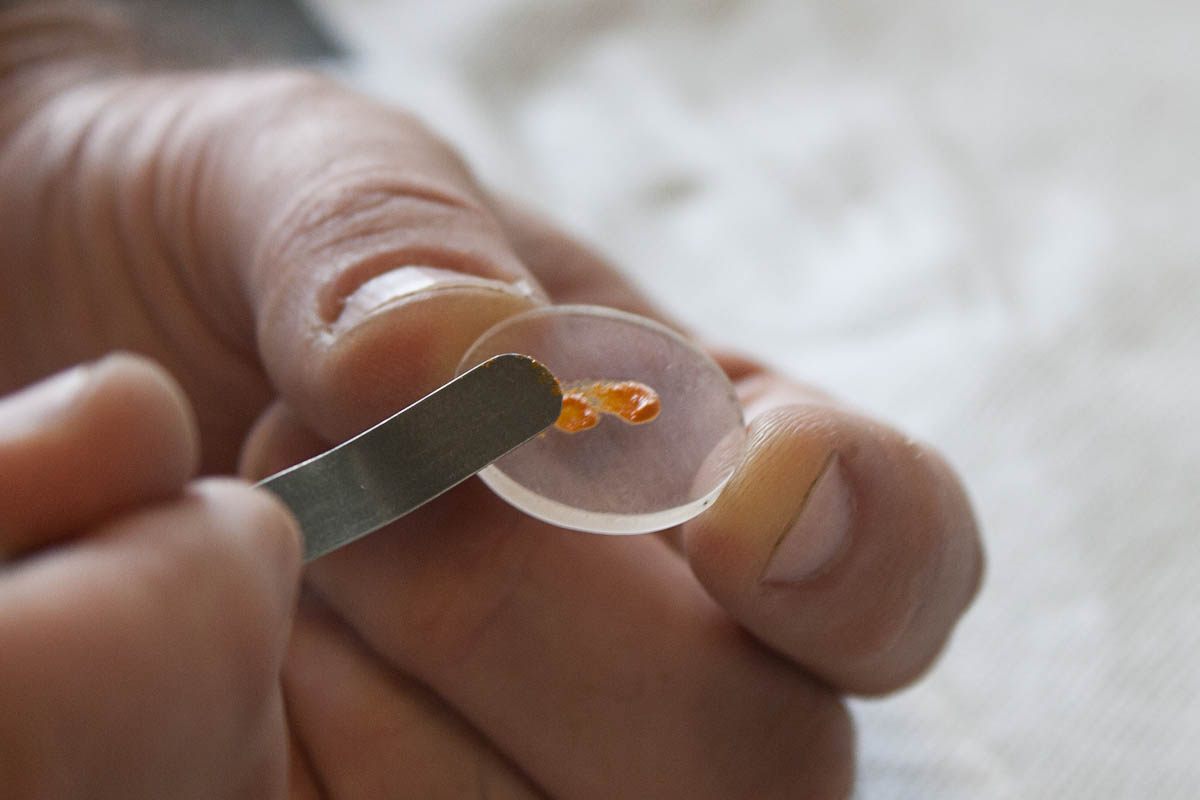
ATR-FTIR has been used in fields spanning forensic analysis to pharmaceutical applications and even art preservation. Due to its ease of use and accessibility ATR can be used to determine the purity of a compound. With only a minimal amount of sample this researcher is able to collect a quick analysis of her sample and determine whether it has been adequately purified or requires further processing. As can be seen in Figure \(\PageIndex{35}\), the sample size is minute and requires no preparation. The sample is placed in close contact with the ATR crystal by turning a knob that will apply pressure to the sample (Figure \(\PageIndex{36}\) ).


ATR has an added advantage in that it inherently encloses the optical path of the IR beam. In a transmission FTIR, atmospheric compounds are constantly exposed to the IR beam and can present significant interference with the sample measurement. Of course the transmission FTIR can be purged in a dry environment, but sample measurement may become cumbersome. In an ATR measurement, however, light from the spectrometer is constantly in contact with the sample and exposure to the environment is reduced to a minimum.
Application to Inorganic Chemistry
One exciting application of ATR is in the study of classical works of art. In the study of fragments of a piece of artwork, where samples are scarce and one-of-a-kind, ATR is a suitable method of characterization because it requires only a small sample size. Determining the compounds present in art enables proper preservation and historical insight into the pieces.
In a study examining several paint samples from a various origins, a micro-ATR was employed for analysis. This study used a silicon crystal with a refractive index of 2.4 and a reduced beam size. Going beyond a simple surface analysis, this study explored the localization of various organic and inorganic compounds in the samples by performing a stratigraphic analysis. The researchers did so by embedding the samples in both KBr and a polyester resins. Two embedding techniques were compared to observe cross-sections of the samples. The mapping of the samples took approximately 1-3 hours which may seem quite laborious to some, but considering the precious nature of the sample, the wait time was acceptable to the researchers.
The optical microscope picture ( Figure \(\PageIndex{37}\) ) shows a sample of a blue painted area from the robe of a 14th century Italian polychrome statue of a Madonna. The spectra shown in Figure \(\PageIndex{38}\) were acquired from the different layers pictured in the box marked in Figure \(\PageIndex{37}\). All spectra were collected from the cross-sectioned sample and the false-color map on each spectrum indicates the location of each of these compounds within the embedded sample. The spectra correspond to the inorganic compounds listed in Table \(\PageIndex{12}\), which also highlights characteristic vibrational bands.

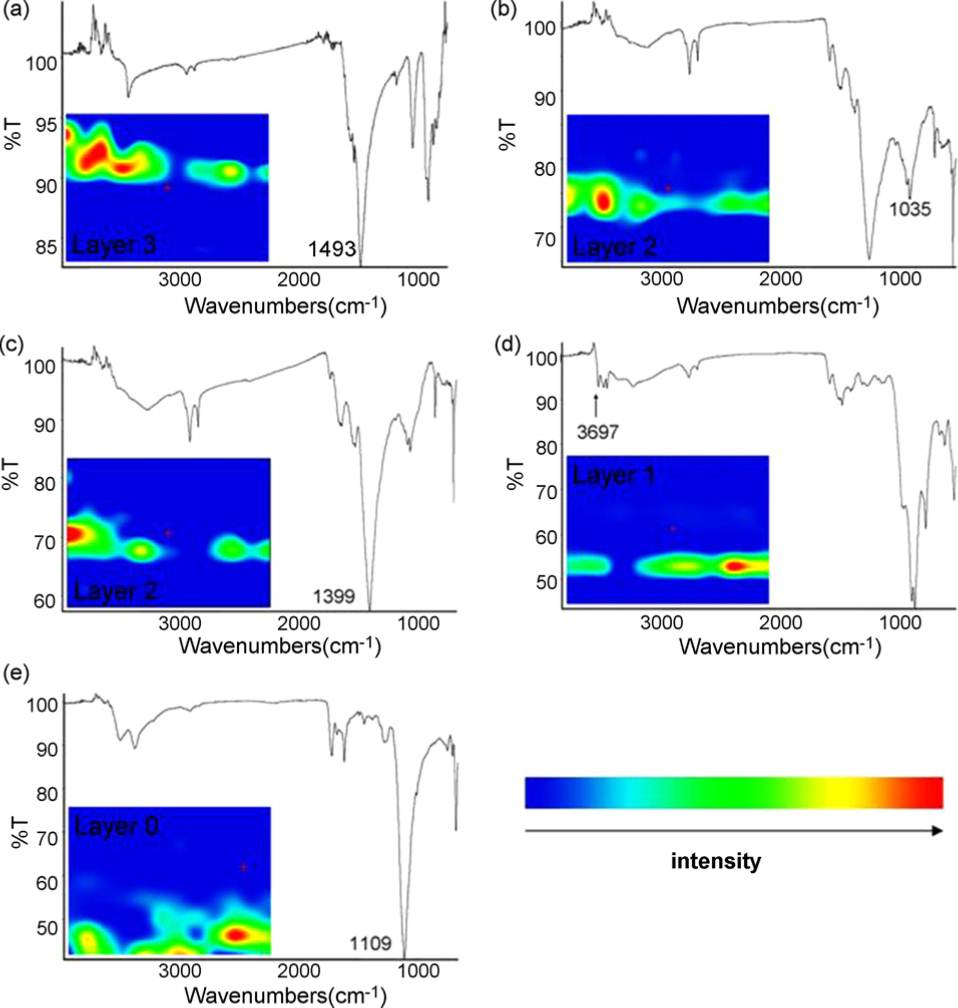
| Compound | Selected Spectral Bands | Assignment |
|---|---|---|
| Cu3(CO3)2(OH)2 (Azurite) | 1493 | CO32- asymmetric stretch |
| Silicate based blue-pigments | 1035 | Si-O stretching |
| 2PbCO3 \(\cdot \) Pb(OH)2 (White lead) | 1399 | CO32- asymmetric stretch |
| A natural ferruginous aluminum silicate red pigment (Bole) | 3697 | OH stretching |
| CaSO4 \(\cdot \) (Gypsum) | 1109 | SO42- asymmetric stretch |
The deep blue layer 3 corresponds to azurite and the light blue paint layer 2 to a mixture of silicate based blue pigments and white lead. Although beyond the ATR crystal’s spatial resolution limit of 20 µm, the absorption of bole was detected by the characteristic triple absorption bands of 3697, 3651, and 3619 cm-1 as seen in spectrum d of Figure \(\PageIndex{37}\). The white layer 0 was identified as gypsum.
To identify the binding material, the KBr embedded sample proved to be more effective than the polyester resin. This was due in part to the overwhelming IR absorbance of gypsum in the same spectral range (1700-1600 cm-1) as a characteristic stretch of the binding as well as some contaminant absorption due to the polyester embedding resin.
To spatially locate specific pigments and binding media, ATR mapping was performed on the area highlighted with a box in Figure \(\PageIndex{37}\). The false color images alongside each spectrum in Figure \(\PageIndex{38}\) indicate the relative presence of the compound corresponding to each spectrum in the boxed area. ATR mapping was achieved by taking 108 spectra across the 220x160 µm area and selecting for each identified compound by its characteristic vibrational band.