
10.4: Photoelectron Spectroscopy
- Page ID
- 13473
- Demonstrate how photoelectron spectroscopy can be used to resolve the absolute energies of molecular orbitals.
Photoelectron spectroscopy (PES) utilizes photo-ionization and analysis of the kinetic energy distribution of the emitted photoelectrons to study the composition and electronic state of the surface region of a sample.
- X-ray Photoelectron Spectroscopy (XPS) uses soft x-rays (with a photon energy of 200-2000 eV) to examine electrons in core-levels.
- Ultraviolet Photoelectron Spectroscopy (UPS) using vacuum UV radiation (with a photon energy of 10-45 eV) to examine electrons in valence levels.
Both photoelectron spectroscopies are based upon a single photon in/electron out process. The energy of a photon of all types of electromagnetic radiation is given by the Planck–Einstein relation:
\[E = h \nu \label{5.3.1} \]
where \(h\) is Planck constant and \(\nu\) is the frequency (Hz) of the radiation. UPS is a powerful technique to exam molecular electron structure since we are interested in the molecular orbitals from polyatomic molecules (especially the valence orbitals) and is the topic of this page.
Photoelectron spectroscopy uses monochromatic sources of radiation (i.e. photons of fixed energy). In UPS the photon interacts with valence levels of the molecule or solid, leading to ionization by removal of one of these valence electrons. The kinetic energy distribution of the emitted photoelectrons (i.e. the number of emitted photoelectrons as a function of their kinetic energy) can be measured using any appropriate electron energy analyzer and a photoelectron spectrum can thus be recorded.

The process of photoionization can be considered in several ways. One way is to look at the overall process for a species \(A\):
\[A + \text{photon} \rightarrow A^+ + e^- \label{5.3.2} \]
Conservation of energy then requires that (after using Equation \(\ref{5.3.1}\)):
\[E(A) + h\nu = E(A^+ ) + E(e^-) \label{5.3.3} \]
Since the free electron's energy is present solely as kinetic energy (\(KE\)) (i.e., there is no internal energy in a free electron)
\[ E(e^-) = KE \nonumber \]
Equation \(\ref{5.3.3}\) can then be rearranged to give the following expression for the KE of the photoelectron:
\[KE = h\nu - \left[ E(A^+ ) - E(A) \right] \label{5.3.4} \]
The final term in brackets represents the difference in energy between the ionized and neutral species and is generally called the vertical ionization energy (\(IE\)) of the ejected electron; this then leads to the following commonly quoted equations:
\[KE = h\nu - IE \label{5.3.5} \]
or
\[IE= h\nu - KE \label{Big} \]
The vertical ionization energy is a direct measure of the energy required to just remove the electron concerned from its initial level to the vacuum level (i.e., a free electron). Photoelectron spectroscopy measures the relative energies of the ground and excited positive ion states that are obtained by removal of single electrons from the neutral molecule.
Equation \ref{5.3.5} may look familiar to you as it the same equation Einstein used to describe the photoelectric effect except the vertical ionization energy (\(IE\)) is substituted for workfunction \(\Phi\). Both vertical ionization energy and workfunctions are metrics for the binding energy of an electron in the sample.
At a fundamental level, ionization energies are well-defined thermodynamic quantities related to the heats of protonation, oxidation/reduction chemistry, and ionic and covalent bond energies. Ionization energies are closely related to the concepts of electronegativity, electron-richness, and the general reactivity of molecules. The energies and other characteristic features of the ionization bands observed in photoelectron spectroscopy provide some of the molecular orbitals detailed and specific quantitative information regarding the electronic structure and bonding in molecules.

Ionization is explicitly defined in terms of transitions between the ground state of a molecule and ion states as shown in Equation \(\ref{Big}\) and as illustrated in the Figure 10.4.2 . Nonetheless, the information obtained from photoelectron spectroscopy is typically discussed in terms of the electronic structure and bonding in the ground states of neutral molecules, with ionization of electrons occurring from bonding molecular orbitals, lone pairs, antibonding molecular orbitals, or atomic cores. These descriptions reflect the relationship of ionization energies to the molecular orbital model of electronic structure.
Ionization energies are directly related to the energies of molecular orbitals by Koopmans' theorem, which states that the negative of the eigenvalue of an occupied orbital from a Hartree-Fock calculation is equal to the vertical ionization energy to the ion state formed by removal of an electron from that orbital (Figure 10.4.3 ), provided the distributions of the remaining electrons do not change (i.e., frozen).
\[ I_j = - \epsilon_j \label{Koopman} \]
There are many limitations to Koopmans' theorem, but in a first order approximation each ionization of a molecule can be considered as removal of an electron from an individual orbital. The ionization energies can then be considered as measures of orbital stabilities, and shifts can be interpreted in terms of orbital stabilizations or destabilizations due to electron distributions and bonding. Koopmans' theorem is implicated whenever an orbital picture is involved, but is not necessary when the focus is on the total electronic states of the positive ions.

Koopmans' theorem argues that the negative of the eigenvalue of an occupied orbital from a Hartree-Fock calculation is equal to the vertical ionization energy to the ion state formed by removal of an electron from that orbital.
Several different ionization energies can be defined, depending on the degree of vibrational excitation of the cations. In general, the following two types of ionization energies are considered (Figure 10.4.4 ):
- Adiabatic ionization energy corresponds to the ionization energy associated with this transition \[M(X, v” = 0) + h\nu \rightarrow M^+(x, v’ = 0) + e^- \nonumber \] Adiabatic ionization energy that is, the minimum energy required to eject an electron from a molecule in its ground vibrational state and transform it into a cation in the lowest vibrational level of an electronic state x of the cation.
- Vertical ionization energy corresponds to the ionization energy associated with this transition \[ M(X, v” = 0) + h\nu \rightarrow M^+(x, v’ = n) + e^- \nonumber \] where, the value n of the vibrational quantum number v’ corresponds to the vibrational level whose wavefunction gives the largest overlap with the v” = 0 wavefunction. This is the most probable transition and usually corresponds to the vertical transition where the internuclear separations of the ionic state are similar to those of the ground state.

The geometry of an ion may be different from the neutral molecule. The measured ionzation energy in a PES experiment can refer to the vertical ionization energy, in which case the ion is in the same geometry as the neutral, or to the adiabatic ionzaiton energy, in which case the ion is in its lowest energy, relaxed geometry (mostly the former though). This is illustrated in the Figure 10.4.4 . For a diatomic the only geometry change possible is the bond length. The figure shows an ion with a slightly longer bond length than the neutral. The harmonic potential energy surfaces are shown in green (neutral) and red (ion) with vibrational energy levels. The vertical ionzation energy is always greater than the adiabatic ionzation energy.
You have been exposed to three metrics of ionization energies already, which are similar, but with distinct differences:
- The ionization energy (also called adiabatic ionization energy) is the lowest energy required to effect the removal of an electron from a molecule or atom, and corresponds to the transition from the lowest electronic, vibrational and rotational level of the isolated molecule to the lowest electronic, vibrational and rotational level of the isolated ion.
- The binding energy (also called vertical ionization energy) is the energy change corresponding to an ionization reaction leading to formation of the ion in a configuration which is the same as that of the equilibrium geometry of the ground state neutral molecule.
- The workfunction is the minimum energy needed to remove an electron from a (bulk) solid to a point in the vacuum.
As you remember, the molecular orbital description of hydrogen involves two \(|1s \rangle\) atomic orbitals generating a bonding \(1\sigma_g\) and antibonding \(2\sigma_u^*\) molecular orbitals. The two electrons that are responsible for the \(\ce{H_2}\) bond are occupied in the \(1\sigma_g\).

The PES spectrum has a single band that corresponds to the ionization of a 1σg electron. The multiple peaks are due to electrons ejecting from a range of stimulated vibrational energy levels. When extensive vibrational structure is resolved in a PES molecular orbital, then the removal of an electron from that molecular orbital induces a significant change in the bonding (in this case an increase in the bond length since the bond order has been reduced).
Diatomic nitrogen is more complex than hydrogen since multiple molecular orbitals are occupied. Four molecular orbitals are occupied (the two \(1\pi_u\) orbitals are both occupied). The UV photoelectron spectrum of \(N_2\), has three bands corresponding to \(3σ_g\), \(1π_u\) and \(2σ_u\) occupied molecular orbitals. Both \(3σ_g\) and \(2σ_u\) are weakly bonding and antibonding. The \(1\sigma_g\) orbital is not resolved in this spectrum since the incident light \(h\nu\) used did not have sufficient energy to ionize electrons in that deeply stabilized molecular orbital.

Note that extensive vibrational structure for the \(1π_u\) band indicates that the removal of an electron from this molecular orbital causes a significant change in the bonding.
Hydrogen Chloride
The molecular energy level diagram for \(\ce{HCl}\) is reproduced in Figure 10.4.5

Important aspects of molecular orbital diagram in Figure 10.4.5 :
- The H 1s energy lies well above the Cl 2s and 2p atomic orbitals;
- The valence electron configuration can be written 3σ21π4;
- The H 1s orbital contributes only to the σ molecular orbitals, as does one of the Cl 2p orbitals (hence the lines in Figure 10.4.5 connecting these atomic orbitals and the 3σ and 4σ molecular orbitals);
- The remaining Cl 2p orbitals (ie those perpendicular to the bond axis) are unaffected by bonding, and these form the 1π molecular orbitals;
- The 1π orbitals are nonbonding - they are not affected energetically by the interaction between the atoms, and are hence neither bonding nor antibonding;
- The 3σ orbital is weakly bonding, and largely Cl 2p;
- The 3σ* orbital is antibonding, and primarily of H 1s character;
Figure 10.4.6 shows the analogous MO diagram and photoelectron spectrum for \(\ce{HCl}\). The spectrum has two bands corresponding to non-bonding 1p (or \(1\pi\)) molecular orbitals (with negligible vibrational structure) and the 3s bonding molecular orbital (vibrational structure).

The higher energy (more stabilized) core molecular orbitals are not observed since the incident photon energy \(h\nu\) is below their ionization energies.
Water
In the simplified valence bond theory perspective of the water molecule, the oxygen atom form four \(sp^3\) hybrid orbitals. Two of these are occupied by the two lone pairs on the oxygen atom, while the other two are used for bonding. Within the molecular orbital picture, the electronic configuration of the \(\ce{H_2O^{+}}\) molecule is \((1a_1)^2 (2a_1)^2 (1b_2)^2 (3a_1)^2 (1b_1)^2\) where the symbols \(a_1\), \(b_2\) and \(b_1\) are orbital labels based on molecular symmetry that will be discussed later (Figure 10.4.7 ). Within Koopmans' theorem:
- The energy of the 1b1 HOMO corresponds to the ionization energy to form the \(\ce{H_2O^{+}}\) ion in its ground state \((1a_1)^2 (2a_1)^2 (1b_2)^2 (3a_1)^2 (1b_1)^1\).
- The energy of the second-highest molecular orbitals \(3a_1\) refers to the ion in the excited state \((1a_1)^2 (2a_1)^2 (1b_2)^2 (3a_1)^1 (1b_1)^2\).

The Hartree–Fock orbital energies (with sign changed) of these orbitals are tabulated below and compared to the experimental ionization energies.
| Molecular orbital | Hartree–Fock orbital Energies (eV) | Experimental Ionization Energies (eV) |
|---|---|---|
| 2a1 | 36.7 | 32.2 |
| 1b2 | 19.5 | 18.5 |
| 3a1 | 15.9 | 14.7 |
| 1b1 | 13.8 | 12.6 |
As explained above, the deviations between orbital energy and ionization energy is small and due to the effects of orbital relaxation as well as differences in electron correlation energy between the molecular and the various ionized states.
The molecular orbital perspective has the lone pair in different orbitals (one in a non-bonding orbital (\(1b_1\) and one in the bonding orbitals). We tern to the photoelectron spectroscopy to help identify which theory is more accurate (i.e., describes reality better). The photoelectron spectrum of water in Figure 10.4.6 can be interpreted as having three major peaks with some fine structure arises from vibrational energy changes. The light source used in this experiment is not sufficiently energetic to ionize electrons from the lowest lying molecular orbitals.
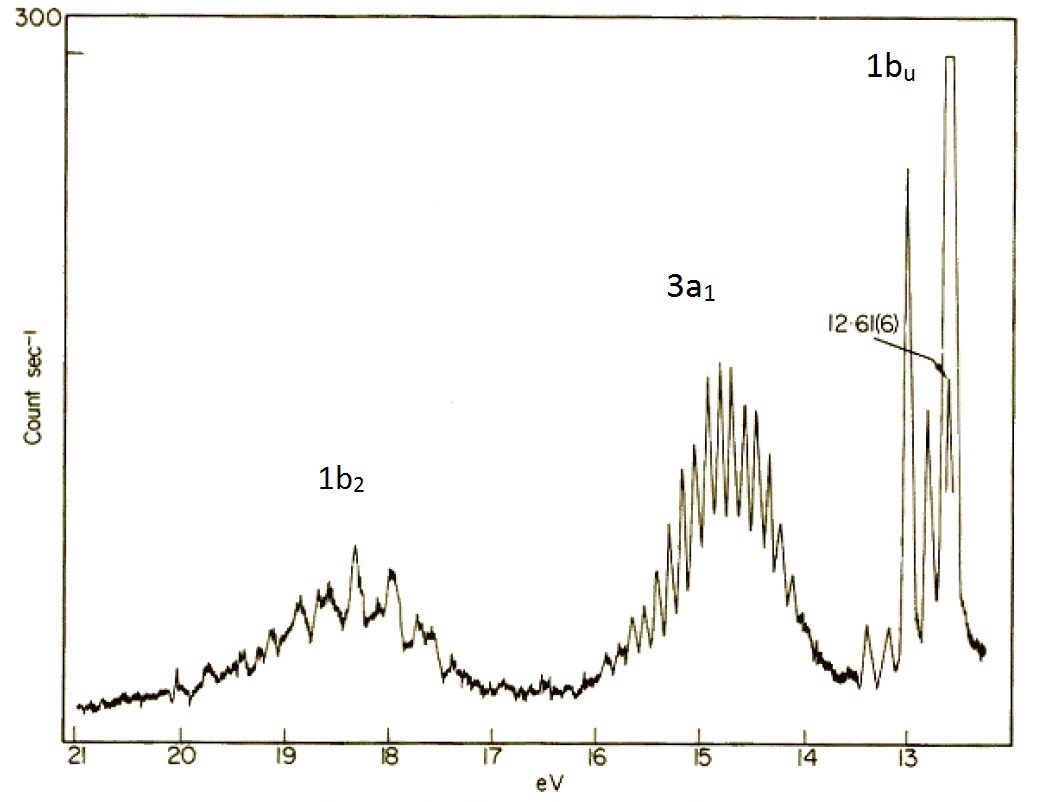
If water was formed two identical O-H bonds and two lone pairs on the oxygen atom line valence bond theory predicts, then the PES in Figure 10.4.8 would have two (degenerate) peaks, one for the two bonds and one for the two lone pairs. The photoelectron spectrum clearly shows three peaks in the positions expected for the molecular orbitals in Figure 10.4.8 .
If the molecular orbitals in Figure 10.4.7 represent the real electronic structure, how do we view the bonding? These molecular orbitals are delocalized and bare little relationship to the familiar 2-center bonds used in valence bond theory. For example, the \(2a_1\) \(1b_1\) and \(3a_1\) molecular orbitals all have contributions from all three atoms, they are really 3-centered molecular orbitals. The bonds however can be thought of as representing a build up of the total electron density which loosely put is a total of all the orbital contributions. Despite this, we keep the ideas of hybridization and 2-center bonds because they are useful NOT because they represent reality
Summary
A photoelecton spectrum can show the relative energies of occupied molecular orbitals by ionization. (i.e. ejection of an electron). A photoelectron spectrum can also be used to determine energy spacing between vibrational levels of a given electronic state. Each orbital energy band has a structure showing ionization to different vibrational levels.
Contributors and Attributions
Roger Nix (Queen Mary, University of London)