13.1: Unit Cells and Crystal Structures
- Page ID
- 396430
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
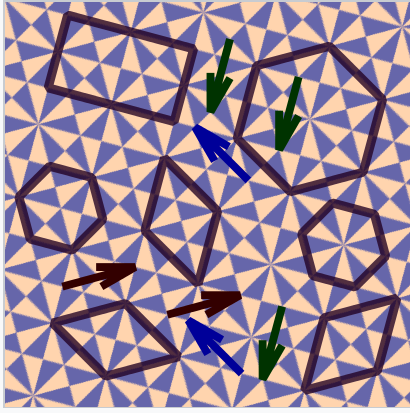
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Crystals can be thought of as repeating patterns, much like wallpaper or bathroom tiles, but in three dimensions. The fundamental repeating unit of the crystal is called the unit cell. It is a three dimensional shape that can be repeated over and over by unit translations to fill space (and leave minimal gaps) in the structure. Some possible unit cells are shown in the tiling pattern at the right, along with arrows that indicate unit translation vectors. In three dimensions, the hexagonal or rhombic unit cells of this pattern would be replaced by three dimensional boxes that would stack together to fill all space. As shown in the figure, the origin of the unit cell is arbitrary. The same set of boxes will fill all space no matter where we define the origin of the lattice. We will see that pure metals typically have very simple crystal structures with cubic or hexagonal unit cells. However the crystal structures of alloys can be quite complicated.
When considering the crystal structures of metals and alloys, it is not sufficient to think of each atom and its neighboring ligands as an isolated system. Instead, think of the entire metallic crystal as a network of atoms connected by a sea of shared valence electrons. The electrons are delocalized because there are not enough of them to fill each "bond" between atoms with an electron pair. For example, in the crystal structures of s-block and p-block metals, each atom has either 8 or 12 nearest neighbors, but the maximum number of s + p electrons is 8. Thus, there are not enough to put two electrons between each pair of atoms. Transition metals can also use their d-orbitals in bonding, but again there are never enough electrons to completely fill all the "bonds."
|
Possible unit cells in a periodic tile pattern. The arrows connect translationally equivalent points (lattice points) in the pattern. |
The atoms in a metal lattice arrange themselves in a certain pattern which can be represented as a 3D box structure known as the unit cell which repeats across the entire metal.
| Simple Cubic | Body Centered Cubic | Face Centered Cubic | Hexagonal Close Packed |
|---|---|---|---|
 |
 |
 |
 |
| 1 atom/cell | 2 atoms/cell | 4 atoms/cell | 2 atoms/cell |
Metal atoms can be approximated as spheres, and therefore are not 100 % efficient in packing, the same way a stack of cannonballs has some empty spaces between the balls. Different unit cells have different packing efficiencies. The number of atoms that is included in the unit cell only includes the fractions of atoms inside of the box. Atoms on the corners of the unit cell count as ⅛ of an atom, atoms on a face count as ½, an atom in the center counts as a full atom. Using this, let's calculate the number of atoms in a simple cubic unit cell, a face centered cubic (fcc) unit cell, and a body centered cubic (bcc) unit cell.
Simple Cubic:
8 corner atoms × ⅛ = 1 atom/cell. The packing in this structure is not efficient (52%) and so this structure type is very rare for metals.
Body Centered Cubic, bcc:
(8 corner atoms × ⅛) + (1 center atom × 1)= 2 atoms/cell. The packing is more efficient (68%) and the structure is a common one for alkali metals and early transition metals. Alloys such as brass (CuZn) also adopt these structures.
Face Centered Cubic, fcc (also called Cubic Close Packed, ccp):
(8 corner atoms × ⅛)+ (6 face atoms × ½)= 4 atoms/cell. This structure, along with its hexagonal relative (hcp), has the most efficient packing (74%). Many metals adopt either the fcc or hcp structure.
Hexagonal Close Packed, hcp:
Like the fcc structure, the packing density of hcp is 74%.
|
The unit cell of a bcc metal contains two atoms. |
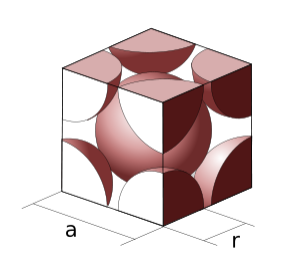
Calculating the packing fraction. The packing fractions of the crystal structures shown above can be calculated by dividing the volume of the atoms in the cell by the volume of the cell itself. The volume of the atoms in the cell is equal to the number of atoms/cell times the volume of a sphere, (4/3)πr3. The volume of the cubic cells is found by cubing the side length. As an example, let's calculate the packing efficiency of a simple cubic unit cell. As we saw earlier in the section, a simple cubic unit cell contains one atom. The side length of the simple cubic unit cell is 2r, since the centers of each atom occupy the corners of the unit cell.
\[\textrm{Packing efficiency} = \frac{(1 \: atom) \times (\frac{4}{3}) \pi r^{3}}{(2r)^{3}}= 0.523\]
The same method can be applied to bcc and fcc structures.
|
Face-centered cubic stack of cannonballs. |