
8.2: Two Mechanistic Models for Nucleophilic Substitution
- Page ID
- 170466

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)As we begin our study of nucleophilic substitution reactions, we will focus at first on simple alkyl halide compounds. While the specific reactions we'll initially consider do not occur in living things, it is nonetheless useful to start with alkyl halides as a model to illustrate some fundamental ideas that we must cover. Later, we will move on to apply what we have earned about alkyl halides to the larger and more complex biomolecules that are undergoing nucleophilic substitution right now in your own cells.
The \(S_N2\) mechanism
You may recall from our brief introduction to the topic in chapter 6 that there are two mechanistic models for how a nucleophilic substitution reaction can proceed. In one mechanism, the reaction is concerted: it takes place in a single step, and bond-forming and bond-breaking occur simultaneously. This is illustrated by the reaction between chloromethane and hydroxide ion:
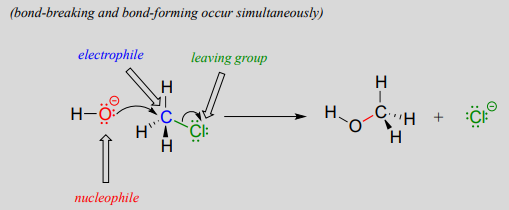
Recall that the hydroxide ion in this reaction is acting as a nucleophile (an electron-rich, nucleus-loving species), the carbon atom of chloromethane is acting as an electrophile (an electron-poor species which is attracted to electrons), and the chloride ion is the leaving group (where the name is self-evident).
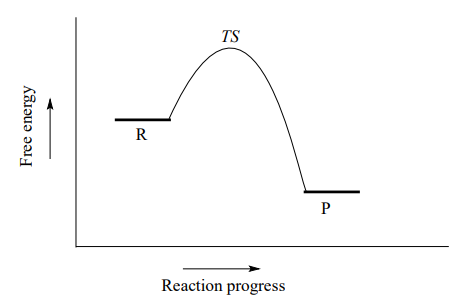
Organic chemists refer to this mechanism by the term '\(S_N2\)', where S stands for 'substitution', the subscript N stands for 'nucleophilic', and the number 2 refers to the fact that this is a bimolecular reaction: the overall rate depends on a step in which two separate species collide. A potential energy diagram for this reaction shows the transition state (TS) as the highest point on the pathway from reactants to products.
The geometry of an \(S_N2\) reaction is specific: the reaction can only occur when the nucleophile collides with the electrophilic carbon from the opposite side relative to the leaving group. This is referred to as backside attack. Approach from the front side simply doesn't work: the leaving group - which, like the nucleophile is an electron-rich group - blocks the way.
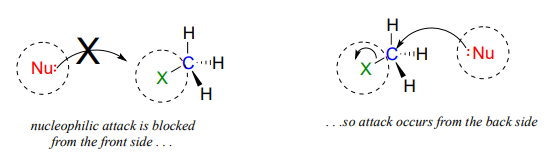 2
2
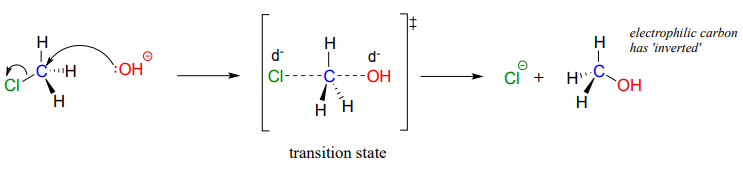
The result of backside attack is that the bonding geometry at the electrophilic carbon inverts (turns inside-out) as the reaction proceeds.
 3
3
The transition state of the reaction is illustrated by drawing dotted lines to represent the covalent bonds that are in the process of breaking or forming. Because the formal charge on the oxygen nucleophile changes from negative one to zero as the reaction proceeds, and conversely the charge on the chlorine leaving group changes from zero to negative one, at the transition state both atoms are shown bearing a partial negative charge (the symbol \(\delta \)-). One other drawing convention for transition states is to use brackets, with the double-dagger symbol in subscript.
Notice that the transition state for an \(S_N2\) reaction has trigonal bipyramidal geometry: the nucleophile, electrophile, and leaving group form a straight line, and the three substituents on carbon (all hydrogen atoms in this case) are arranged in the same plane at \(120^{\circ}\) angles.
What is the measure in degrees) of the \(H-C-O\) angle in the \(S_N2\) transition state illustrated above?
Consider what would happen if we were to replace one of the hydrogen atoms in chloromethane with deuterium (the \(^2H\) isotope), and one with tritium (the radioactive \(^3H\) isotope). Now, because it has four different substituents, our carbon electrophile is a chiral center. We'll arbitrarily assume that we start with the \(S\) enantiomer.
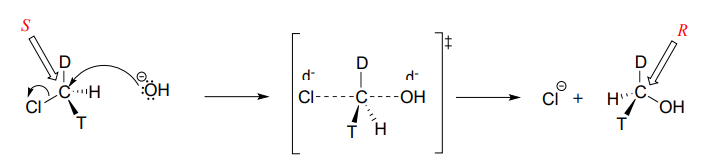
As the hydroxide nucleophile attacks from the backside and the bonding geometry at carbon inverts, we see that the stereochemistry of the product reflects this inversion: we end up with the \(R\) enantiomer of the chiral product.
\(S_N2\) reactions proceed with inversion of stereochemical configuration at the electrophilic carbon.
video tutorial/animation: inversion of configuration during SN2 reactions
The \(S_N1\) mechanism
A second model for the nucleophilic substitution reaction is called the \(S_N1\) mechanism. The '1' in \(S_N1\) indicates that the rate-determining step of the reaction is unimolecular: in other words, the rate-determining step involves a single molecule breaking apart (rather than two molecules colliding as was the case in the \(S_N2\) mechanism.)
In an \(S_N1\) mechanism the carbon-leaving group bond breaks first, before the nucleophile approaches, resulting in formation of a carbocation intermediate (step 1):
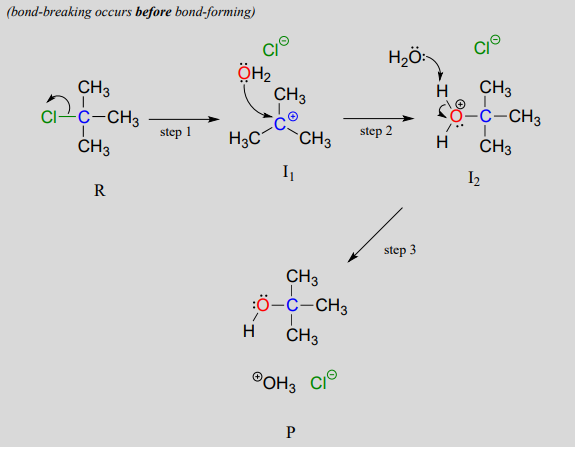
A carbocation is a powerful electrophile: because the carbon lacks a complete octet of valence electrons, it is 'electron-hungry'. In step 2, a lone pair of electrons on the water nucleophile fills the empty p orbital of the carbocation to form a new bond.
Notice that this is actually a three-step mechanism, with a final, rapid acid-base step leading to the alcohol product.
A potential energy diagram for this \(S_N1\) reaction shows that each of the two positively-charged intermediate stages (\(I_1\) and \(I_2\) in the diagram) can be visualized as a valley in the path of the reaction, higher in energy than both the reactant and product but lower in energy than the transition states.
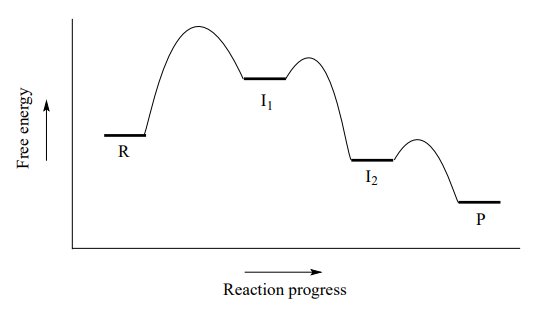
The first, bond-breaking step is the slowest, rate-determining step - notice it has the highest activation energy and leads to the highest-energy species (\(I_1\), the carbocation intermediate). Step 2 is rapid: a new covalent bond forms between a carbocation and and a water nucleophile, and no covalent bonds are broken. Recall from chapter 7 that Bronsted-Lowry proton transfer steps like step 3 are rapid, with low activation energies.
The nucleophilic substitution reactions we have seen so far are examples of hydrolysis. This term is one that you will encounter frequently in organic and biological chemistry. Hydrolysis means 'breaking with water': in a hydrolysis reaction, a water molecule (or hydroxide ion) participates in the breaking of a covalent bond. There are many reaction types other than nucleophilic substitution that can accurately be described as hydrolysis, and we will see several examples throughout the remaining chapters of this book.
Solvolysis is a more general term, used when a bond in a reagent is broken by a solvent molecule: usually, the solvent in question is water or an alcohol such as methanol or ethanol.
Draw a mechanism for the \(S_N1\) solvolysis of tert-butyl chloride in methanol. What new functional group has been formed?
We saw that \(S_N2\) reactions result in inversion of stereochemical configuration at the carbon center. What about the stereochemical outcome of \(S_N1\) reactions? Recall that a carbocation is \(sp^2\)-hybridized, with an empty p orbital perpendicular to the plane formed by the three sigma bonds:
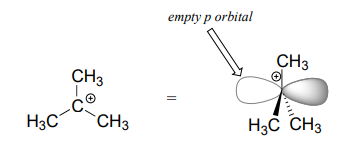
In the second step of an \(S_N1\) reaction, the nucleophile can attack from either side of the carbocation (the leaving group is already gone, and thus cannot block attack from one side like in an \(S_N2\) reaction).
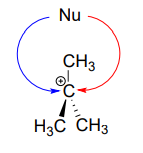
Consider an \(S_N1\) reaction with a chiral tertiary alkyl chloride:
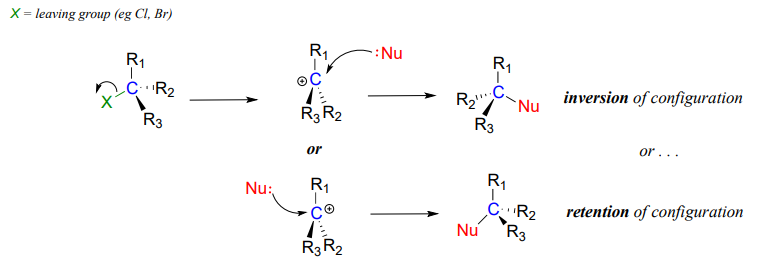
Because the nucleophile is free to attack from either side of the carbocation electrophile, the reaction leads to a 50:50 mixture of two stereoisomeric products. In other words: In general nonenzymatic SN1 reaction can occur with either retention or inversion of configuration at the electrophilic carbon, leading to racemization if the carbon is chiral.
For an example, consider the hydrolysis of (S)-3-chloro-3-methylhexane.
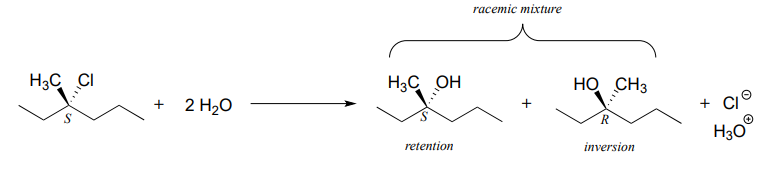
The result of this (nonenzymatic) reaction is a racemic mixture of chiral alcohols.
It is important to remember, however, that enzymatic reactions are in almost all cases very specific with regard to stereochemical outcome. A biochemical \(S_N1\) reaction, as we shall see later, can result in either inversion or retention of configuration at the electrophilic carbon, but generally not a mixture of both: the two reactants are bound with specific geometry in the enzyme's active site, so that the nucleophile can approach from one side only.
(The following exercises refer to nonbiological reactions)
- Draw a complete mechanism for the hydrolysis reaction in the previous figure, showing all bond-breaking and bond-forming steps, and all intermediate species.
- Draw structures representing TS1 and TS2 in the reaction. Use the solid/dash wedge convention to show three dimensions.
- What is the expected optical rotation of the product mixture?
- Could the two organic products be separated on a silica column chromatography?
- Draw the product(s) of the hydrolysis of (R)-3-chloro-3-methyl heptane.
- What can you predict, if anything, about the optical rotation of the product(s)?
- Draw the product(s) of the hydrolysis of (3R,5R)-3-chloro-3,5-dimethyl heptane.
- What can you predict, if anything, about the optical rotation of the product(s)?
Before we go on to look at some actual biochemical nucleophilic substitution reactions, we first need to lay the intellectual groundwork by focusing more closely on the characteristics of the three principal partners in the nucleophilic substitution reaction: the nucleophile, the electrophile, and the leaving group. In addition, we need to consider the carbocation intermediate that plays such a key role in the \(S_N1\) mechanism. For the sake of simplicity, we will continue to use simple, non-biological organic molecules and reaction examples as we work through the basic concepts.
Contributors
Organic Chemistry With a Biological Emphasis by Tim Soderberg (University of Minnesota, Morris)